KAT7 Is a Therapeutic Vulnerability of MLL-Rearranged Acute Myeloid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -

DIPPER, a Spatiotemporal Proteomics Atlas of Human Intervertebral Discs
TOOLS AND RESOURCES DIPPER, a spatiotemporal proteomics atlas of human intervertebral discs for exploring ageing and degeneration dynamics Vivian Tam1,2†, Peikai Chen1†‡, Anita Yee1, Nestor Solis3, Theo Klein3§, Mateusz Kudelko1, Rakesh Sharma4, Wilson CW Chan1,2,5, Christopher M Overall3, Lisbet Haglund6, Pak C Sham7, Kathryn Song Eng Cheah1, Danny Chan1,2* 1School of Biomedical Sciences, , The University of Hong Kong, Hong Kong; 2The University of Hong Kong Shenzhen of Research Institute and Innovation (HKU-SIRI), Shenzhen, China; 3Centre for Blood Research, Faculty of Dentistry, University of British Columbia, Vancouver, Canada; 4Proteomics and Metabolomics Core Facility, The University of Hong Kong, Hong Kong; 5Department of Orthopaedics Surgery and Traumatology, HKU-Shenzhen Hospital, Shenzhen, China; 6Department of Surgery, McGill University, Montreal, Canada; 7Centre for PanorOmic Sciences (CPOS), The University of Hong Kong, Hong Kong Abstract The spatiotemporal proteome of the intervertebral disc (IVD) underpins its integrity *For correspondence: and function. We present DIPPER, a deep and comprehensive IVD proteomic resource comprising [email protected] 94 genome-wide profiles from 17 individuals. To begin with, protein modules defining key †These authors contributed directional trends spanning the lateral and anteroposterior axes were derived from high-resolution equally to this work spatial proteomes of intact young cadaveric lumbar IVDs. They revealed novel region-specific Present address: ‡Department profiles of regulatory activities -

Comprehensive Analysis of Expression and Prognostic Value for JMJD5 and PKM2 in Stomach Adenocarcinoma
Comprehensive Analysis of Expression and Prognostic Value for JMJD5 and PKM2 in Stomach Adenocarcinoma Ling Qi Tumor Hospital of Harbin Medical University Li-Sha Li Tumor Hospital of Harbin Medical University Chao Zhan Tumor Hospital of Harbin Medical University Ming-Xia Jiang Tumor Hospital of Harbin Medical University Dong-Feng Song Tumor Hospital of Harbin Medical University Yi-Ming Wu Tumor Hospital of Harbin Medical University Jun-Qing Gan Tumor Hospital of Harbin Medical University Ke-Xin Jin Tumor Hospital of Harbin Medical University Mei Huang Tumor Hospital of Harbin Medical University yanjing Li ( [email protected] ) Tumor Hospital of Harbin Medical University https://orcid.org/0000-0003-0174-2017 Xiao-Xue Du Tumor Hospital of Harbin Medical University Cheng-Xin Song Tumor Hospital of Harbin Medical University Primary research Keywords: JMJD family, JMJD5, PKM2, stomach adenocarcinoma, bioinformatics analysis Posted Date: June 7th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-569965/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Jumonji C-domain-containing (JMJD) family, a group of genes that regulate epigenetics, is involved in tumor development in several types of cancer. JMJD5 is a member of the JMJD family, and its clinical impact on stomach adenocarcinoma (STAD) remains unclear. Pyruvate kinase M2 (PKM2) promotes metabolism, tumor proliferation, and metastasis in various cancer types. However, the relationship between JMJD5 and PKM2 in STAD is yet to be established. In this study, we investigated the expressions and relationship of JMJD5 and PKM2 in patients with STAD. -

Polymorphisms in GP6, PEAR1A, MRVI1, PIK3CG, JMJD1C, and SHH Genes in Patients with Unstable Angina
International Journal of Environmental Research and Public Health Article Polymorphisms in GP6, PEAR1A, MRVI1, PIK3CG, JMJD1C, and SHH Genes in Patients with Unstable Angina Rafał Rudzik 1, Violetta Dziedziejko 2, Monika Ewa Ra´c 2 , Marek Sawczuk 3, Agnieszka Maciejewska-Skrendo 4 , Krzysztof Safranow 2 and Andrzej Pawlik 1,* 1 Department of Physiology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] 2 Department of Biochemistry and Medical Chemistry, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] (V.D.); [email protected] (M.E.R.); [email protected] (K.S.) 3 Insitute of Physical Culture Sciences, University of Szczecin, 70-111 Szczecin, Poland; [email protected] 4 Faculty of Physical Culture, Gdansk University of Physical Education and Sport, 80-336 Gdansk, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-91-466-1611 Received: 16 September 2020; Accepted: 13 October 2020; Published: 15 October 2020 Abstract: Introduction: Coronary artery disease (CAD) is a significant public health problem because it is one of the major causes of death worldwide. Several studies have investigated the associations between CAD and polymorphisms in genes connected with platelet aggregation and the risk of venous thromboembolism. Aim: In this study, we examined the associations between polymorphisms in GP6 (rs1671152), PEAR1A (rs12566888), MRVI1 (rs7940646), PIK3CG (rs342286), JMJD1C (rs10761741), SHH (rs2363910), and CAD in the form of unstable angina as well as selected clinical and biochemical parameters. The study enrolled 246 patients with diagnosed unstable angina and 189 healthy controls. Results: There were no significant differences in the distribution of the studied polymorphisms between the patients with unstable angina and the controls. -

Epigenetic Regulation of Acute Myeloid Leukaemic Stem Cells
Epigenetic Regulation of Acute Myeloid Leukaemic Stem Cells Halina Hoi Laam Leung Bachelor of Medicinal Chemistry Advanced (Honours Class 1) A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Children‘s Cancer Institute Australia for Medical Research School of Women‘s and Children‘s Health Faculty of Medicine University of New South Wales November 2015 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Leung First name: Halina Other name/s: Hoi Laam Abbreviation for degree as given in the University calendar: PhD School: Women’s and Children’s Health Faculty: Medicine Title: Epigenetic Regulation of Acute Myeloid Leukaemic Stem Cells Abstract 350 words maximum: (PLEASE TYPE) Acute myeloid leukaemia (AML) is a heterogeneous blood disease with high relapse rates. A small population of highly drug resistant leukaemic stem cells (LSC) persists after chemotherapy, causing disease relapse. This emphasises the need to develop novel LSC-targeted therapies. While increasing evidence has emerged in recent years highlighting the role of epigenetic regulators in cancer development and maintenance, knowledge in epigenetic regulation of LSC is limited. This study investigated the role of two novel histone demethylases, Jmjd1c and Jmjd5, in regulating LSC, as well as the effectiveness of currently available epigenetic agents on leukaemogenesis. This study has shown that Jmjd1c is required for AML development and maintenance. Experiments involving enforced expression of Jmjd1c in haematopoietic stem cells (HSC) suggested that Jmjd1c has the capacity to enhance stem cell proliferation. Jmjd1c overexpression also promoted proliferation of HSC transduced with oncogenes, Hoxa9/Meis1a, and accelerated leukaemia development in mice. -

Multipronged Approach to Study Glaucoma-Associated Phenotypes
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 8-2016 Multipronged Approach to Study Glaucoma- Associated Phenotypes Sumana Rameshbabu Chintalapudi University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Neurosciences Commons Recommended Citation Chintalapudi, Sumana Rameshbabu (http://orcid.org/0000-0003-1079-0950), "Multipronged Approach to Study Glaucoma- Associated Phenotypes" (2016). Theses and Dissertations (ETD). Paper 404. http://dx.doi.org/10.21007/etd.cghs.2016.0410. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Multipronged Approach to Study Glaucoma-Associated Phenotypes Document Type Dissertation Degree Name Doctor of Philosophy (PhD) Program Integrated Program in Biomedical Sciences Track Neuroscience Research Advisor Monica M. Jablonski, Ph.D. Committee Edward Chaum, M.D., Ph.D. Dianna A. Johnson, Ph.D. Rajendra Raghow, Ph.D. Vanessa Morales-Tirado, Ph.D. Robert W. Williams, Ph.D. ORCID http://orcid.org/0000-0003-1079-0950 DOI 10.21007/etd.cghs.2016.0410 Comments One year embargo expires August 2017. This dissertation is available at UTHSC Digital Commons: https://dc.uthsc.edu/dissertations/404 Multipronged Approach to Study Glaucoma-Associated Phenotypes A Dissertation Presented for The Graduate Studies Council The University of Tennessee Health Science Center In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy From The University of Tennessee By Sumana Rameshbabu Chintalapudi August 2016 . -

Identification and Characterization of the TRIP8 and REEP3 Genes on Chromosome 10Q21.3 As Novel Candidate Genes for Autism
European Journal of Human Genetics (2007) 15, 422–431 & 2007 Nature Publishing Group All rights reserved 1018-4813/07 $30.00 www.nature.com/ejhg ARTICLE Identification and characterization of the TRIP8 and REEP3 genes on chromosome 10q21.3 as novel candidate genes for autism Dries Castermans1, Joris R Vermeesch2, Jean-Pierre Fryns2, Jean G Steyaert2,4,5, Wim J M Van de Ven3, John W M Creemers1 and Koen Devriendt*,2 1Laboratory for Biochemical Neuroendocrinology, Department of Human Genetics, Flanders Interuniversity Institute for Biotechnology, Catholic University of Leuven, Leuven, Belgium; 2Centre for Human Genetics, Division of Clinical Genetics, Catholic University of Leuven, Leuven, Belgium; 3Laboratory for Molecular Oncology, Department of Human Genetics, Flanders Interuniversity Institute for Biotechnology, Catholic University of Leuven, Leuven, Belgium; 4Department of Child Psychiatry, Catholic University of Leuven, Leuven, Belgium; 5Centre for Clinical Genetics, University of Maastricht, Maastricht, The Netherlands Autism is a genetic neurodevelopmental disorder of unknown cause and pathogenesis. The identification of genes involved in autism is expected to increase our understanding of its pathogenesis. Infrequently, neurodevelopmental disorders like autism are associated with chromosomal anomalies. To identify candidate genes for autism, we initiated a positional cloning strategy starting from individuals with idiopathic autism carrying a de novo chromosomal anomaly. We report on the clinical, cytogenetic and molecular findings in a male person with autism, no physical abnormalities and normal IQ, carrying a de novo balanced paracentric inversion 46,XY,inv(10)(q11.1;q21.3). The distal breakpoint disrupts the TRIP8 gene, which codes for a protein predicted to be a transcriptional regulator associated with nuclear thyroid hormone receptors. -

JMJD1C Is Required for the Survival of Acute Myeloid Leukemia by Functioning As a Coactivator for Key Transcription Factors
Downloaded from genesdev.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors Mo Chen,1 Nan Zhu,2,6 Xiaochuan Liu,3 Benoit Laurent,4,5 Zhanyun Tang,1 Rowena Eng,2 Yang Shi,4,5 Scott A. Armstrong,2 and Robert G. Roeder1 1Laboratory of Biochemistry and Molecular Biology, The Rockefeller University, New York, New York 10065, USA; 2Human Oncology and Pathogenesis Program, Memorial Sloan-Kettering Cancer Center, New York, New York 10065, USA; 3Department of Microbiology, Biochemistry, and Molecular Genetics, Rutgers University, Newark, New Jersey 07103, USA; 4Division of Newborn Medicine, Epigenetics Program, Department of Medicine, Boston Children’s Hospital, Boston, Massachusetts 02115, USA; 5Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, USA RUNX1–RUNX1T1 (formerly AML1-ETO), a transcription factor generated by the t(8;21) translocation in acute myeloid leukemia (AML), dictates a leukemic program by increasing self-renewal and inhibiting differentiation. Here we demonstrate that the histone demethylase JMJD1C functions as a coactivator for RUNX1–RUNX1T1 and is required for its transcriptional program. JMJD1C is directly recruited by RUNX1–RUNX1T1 to its target genes and regulates their expression by maintaining low H3K9 dimethyl (H3K9me2) levels. Analyses in JMJD1C knockout mice also establish a JMJD1C requirement for RUNX1–RUNX1T1’s ability to increase proliferation. We also show a critical role for JMJD1C in the survival of multiple human AML cell lines, suggesting that it is required for leukemic programs in different AML cell types through its association with key transcription factors. -
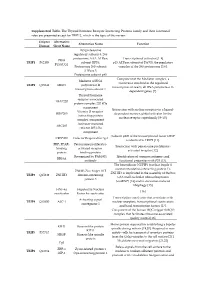
Supplemental Table. the Thyroid Hormone Receptor Interacting
Supplemental Table. The Thyroid hormone Receptor Interacting Proteins family and their functional roles are presented except for TRIP12, which is the topic of this review. Uniprot Alternative Alternative Name Function Human Short Name 26S proteasome regulatory subunit 8, 26S proteasome AAA-ATPase Transcriptional activator [1–4] PRS8 TRIP1 P62195 subunit RPT6, p45 ATPase subunit of PA700, the regulatory P45/SUG1 Proteasome 26S subunit complex of the 26S proteasome [5,6]. ATPase 5. Proteasome subunit p45 Component of the Mediator complex, a Mediator of RNA coactivator involved in the regulated TRIP2 Q15648 MED1 polymerase II transcription of nearly all RNA polymerase II- transcription subunit 1 dependent genes [7]. Thyroid hormone receptor-associated TRAP220 protein complex 220 kDa component Interaction with nuclear receptors in a ligand- Vitamin D receptor- DRIP205 dependent manner, global activator for the interacting protein nuclear receptor superfamily [8–10]. complex component Activator-recruited ARC205 cofactor 205 kDa component Subunit p200 of the transcriptional factor CRSP CRSP200 Cofactor Required for Sp1 is identical to TRIP2 [11]. PBP, PPAR- Peroxisome proliferator- Interaction with peroxisome proliferator- binding activated receptor- activated receptor [12]. protein binding protein Recognized by PAb1801 Identification of common antigenic and RB18A antibody functional properties with P53 [13]. The heterodimer NUFIP1 (nuclear fragile X mental retardation-interacting protein 1)- ZNHI3 Zinc finger HIT ZNHIT3 is implicated in the assembly of the box TRIP3 Q15649 ZNHIT3 domain-containing C/D small nucleolar ribonucleoprotein protein 3 (snoRNP) [14] and in starvation-induced ribophagy [15]. HNF-4a Hepatocyte Nuclear [16] coactivator Factor 4a coactivator Transcription coactivator that associates with Activating signal TRIP4 Q15650 ASC-1 nuclear receptors, transcriptional coactivators cointegrator 1 and basal transcription factors [17]. -

Integrative Genomic Analyses on Ikaros and Its Expression Related to Solid Cancer Prognosis
571-577.qxd 24/6/2010 12:45 ÌÌ ™ÂÏ›‰·571 ONCOLOGY REPORTS 24: 571-577, 2010 571 Integrative genomic analyses on Ikaros and its expression related to solid cancer prognosis LIMING YANG1, YUMING LUO1 and JIFU WEI2 1School of Life Sciences, Huaiyin Normal University, 111 Changjiang West Road, Huai'an 223300; 2Clinical Research Center, the First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, P.R. China Received March 10, 2010; Accepted May 3, 2010 DOI: 10.3892/or_00000894 Abstract. Ikaros is a member of the Kruppel family of zinc Introduction finger DNA-binding proteins. The Ikaros protein contains two separate regions of zinc-finger domains: 4 DNA-binding zinc The Ikaros (IKZF1, Lyf-1) is a member of the Kruppel fingers near the N-terminus and 2 zinc fingers for protein- family of zinc finger DNA-binding proteins 1 (1). The Ikaros protein interactions near the C-terminus. Here, we identified protein contains two separate regions of zinc-finger domains: the Ikaros gene from 14 vertebrate genomes and found Ikaros 4 DNA-binding zinc fingers near the N-terminus and 2 zinc existed in all kinds of vertebrate including fish, amphibians, fingers for protein-protein interactions near the C-terminus. birds and mammals. Moreover, except rat and Xenopus The human Ikaros gene, located at 7p12, contains seven exons tropicalis Ikaros proteins, which lack the first C2H2-type 1 and gives rise to at least eight isoforms by alternative splicing Zinc finger region, all identified Ikaros proteins contain six (2). All isoforms share a common C-terminal domain that C2H2-type 1 Zinc finger regions. -

Comparative Integromics on JMJD1C Gene Encoding Histone Demethylase
219-223 30/5/07 15:19 Page 219 INTERNATIONAL JOURNAL OF ONCOLOGY 31: 219-223, 2007 219 Comparative integromics on JMJD1C gene encoding histone demethylase: Conserved POU5F1 binding site elucidating mechanism of JMJD1C expression in undifferentiated ES cells and diffuse-type gastric cancer MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center Research Institute, Tokyo 104-0045, Japan Received March 22, 2007; Accepted April 30, 2007 Abstract. Epigenetic modifications of genomic DNA and analyses revealed that binding sites for POU5F1 (OCT3/ histones alter the chromatin structure to regulate the accessi- OCT4), AP-1, and bHLH transcription factors within the bility of transcription factors to the promoter or enhancer promoter region located 5' to exon 1B of human JMJD1C regions. In 2003, we identified and characterized JMJD1C gene were conserved in chimpanzee, cow, mouse and rat (TRIP8) consisting of TRI8H1 domain with C2HC4-type JMJD1C orthologs. POU5F1-mediated expression of JMJD1C zinc finger-like motif, TRI8H2 domain with thyroid hormone histone demethylase is implicated in the reactivation of receptor ß-binding region, and JmjC domain. JMJD1A (TSGA), silenced genes in undifferentiated ES cells, pancreatic islet, JMJD1B (5qNCA) and JMJD1C with the common domain and diffuse-type gastric cancer. architecture are histone H3K9 demethylases implicated in the nuclear hormone receptor-based transcriptional regulation. Introduction Here, comparative integromics on JMJD1C gene is reported. JMJD1C variant 1, previously reported, consists of exons 1, Embryogenesis generating whole body from fertilized egg in 2 and 3-26, while JMJD1C variant 2 characterized in this the cell autonomous manner is regulated by the network of study was transcribed from novel exon 1B located 5' to exon transcription factors as well as by the epigenetic modifications 3. -
Crucial Functions of the JMJD1/KDM3 Epigenetic Regulators in Cancer Yuan Sui1, Ruicai Gu2, and Ralf Janknecht1,2,3
Published OnlineFirst June 30, 2020; DOI: 10.1158/1541-7786.MCR-20-0404 MOLECULAR CANCER RESEARCH | REVIEW Crucial Functions of the JMJD1/KDM3 Epigenetic Regulators in Cancer Yuan Sui1, Ruicai Gu2, and Ralf Janknecht1,2,3 ABSTRACT ◥ Epigenetic changes are one underlying cause for cancer devel- invasion, and facilitate efficient tumor growth. However, JMJD1A opment and often due to dysregulation of enzymes modifying DNA may also inhibit the formation of germ cell tumors. Likewise, or histones. Most Jumonji C domain-containing (JMJD) proteins JMJD1B appears to be a tumor suppressor in acute myeloid are histone lysine demethylases (KDM) and therefore epigenetic leukemia, but a tumor promoter in other cancers. Notably, by regulators. One JMJD subfamily consists of JMJD1A/KDM3A, reducing methylation levels on histone H3 lysine 9, JMJD1 proteins JMJD1B/KDM3B, and JMJD1C/KDM3C that are roughly 50% can profoundly alter the transcriptome and thereby affect tumor- identical at the amino acid level. All three JMJD1 proteins are igenesis, including through upregulating oncogenes such as capable of removing dimethyl and monomethyl marks from lysine 9 CCND1, JUN, and MYC. This epigenetic activity of JMJD1 proteins on histone H3 and might also demethylate histone H4 on arginine 3 is sensitive to heavy metals, oncometabolites, oxygen, and reactive and nonhistone proteins. Analysis of knockout mice revealed oxygen species, whose levels are frequently altered within cancer critical roles for JMJD1 proteins in fertility, obesity, metabolic cells. In conclusion, inhibition of JMJD1 enzymatic activity through syndrome, and heart disease. Importantly, a plethora of studies small molecules is predicted to be beneficial in many different demonstrated that especially JMJD1A and JMJD1C are overex- cancers, but not in the few malignancies where JMJD1 proteins pressed in various tumors, stimulate cancer cell proliferation and apparently exert tumor-suppressive functions.