The Ketamine Antidepressant Story: New Insights
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
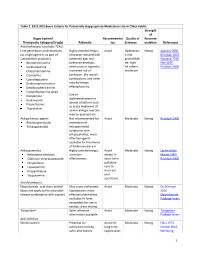
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Involvement of Catecholaminergic and Gabaaergic Mediations in the Anxiety-Related Behavior in Long-Term Powdered Diet-Fed Mice T
Neurochemistry International 124 (2019) 1–9 Contents lists available at ScienceDirect Neurochemistry International journal homepage: www.elsevier.com/locate/neuint Involvement of catecholaminergic and GABAAergic mediations in the anxiety-related behavior in long-term powdered diet-fed mice T ∗ Fukie Yaoitaa, , Masahiro Tsuchiyab, Yuichiro Araic, Takeshi Tadanod, Koichi Tan-Noa a Department of Pharmacology, Faculty of Pharmaceutical Sciences, Tohoku Medical and Pharmaceutical University, 4-4-1 Komatsushima, Aoba-ku, Sendai, 981-8558, Japan b Department of Nursing, Tohoku Fukushi University, 1-8-1 Kunimi, Aoba-ku, Sendai, 981-8522, Japan c Tokyo Ariake University of Medical and Health Science, 2-9-1 Ariake, Koto-Ku, Tokyo, 135-0063, Japan d Complementary and Alternative Medicine Clinical Research and Development, Graduate School of Medicine Sciences, Kanazawa University, Kakumamachi, Kanazawa, 920-1192, Japan ARTICLE INFO ABSTRACT Keywords: Dietary habits are important factors which affect metabolic homeostasis and the development of emotion. We Atomoxetine have previously shown that long-term powdered diet feeding in mice increases spontaneous locomotor activity Methylphenidate and social interaction (SI) time. Moreover, that diet causes changes in the dopaminergic system, especially PD168077 increased dopamine turnover and decreased dopamine D4 receptor signals in the frontal cortex. Although the Anxiety-related behavior increased SI time indicates low anxiety, the elevated plus maze (EPM) test shows anxiety-related behavior and Low anxiety impulsive behavior. In this study, we investigated whether the powdered diet feeding causes changes in anxiety- Bicuculline Attention deficit/hyperactivity disorder related behavior. Mice fed a powdered diet for 17 weeks from weaning were compared with mice fed a standard diet (control). -

Citizen Cyborg.” Citizen a Groundbreaking Work of Social Commentary, Citizen Cyborg Artificial Intelligence, Nanotechnology, and Genetic Engineering —DR
hughes (continued from front flap) $26.95 US ADVANCE PRAISE FOR ARTIFICIAL INTELLIGENCE NANOTECHNOLOGY GENETIC ENGINEERING MEDICAL ETHICS INVITRO FERTILIZATION STEM-CELL RESEARCH $37.95 CAN citizen LIFE EXTENSION GENETIC PATENTS HUMAN GENETIC ENGINEERING CLONING SEX SELECTION ASSISTED SUICIDE UNIVERSAL HEALTHCARE human genetic engineering, sex selection, drugs, and assisted In the next fifty years, life spans will extend well beyond a century. suicide—and concludes with a concrete political agenda for pro- cyborg Our senses and cognition will be enhanced. We will have greater technology progressives, including expanding and deepening control over our emotions and memory. Our bodies and brains “A challenging and provocative look at the intersection of human self-modification and human rights, reforming genetic patent laws, and providing SOCIETIES MUST RESPOND TO THE REDESIGNED HUMAN OF FUTURE WHY DEMOCRATIC will be surrounded by and merged with computer power. The limits political governance. Everyone wondering how society will be able to handle the coming citizen everyone with healthcare and a basic guaranteed income. of the human body will be transcended, as technologies such as possibilities of A.I. and genomics should read Citizen Cyborg.” citizen A groundbreaking work of social commentary, Citizen Cyborg artificial intelligence, nanotechnology, and genetic engineering —DR. GREGORY STOCK, author of Redesigning Humans illuminates the technologies that are pushing the boundaries of converge and accelerate. With them, we will redesign ourselves and humanness—and the debate that may determine the future of the our children into varieties of posthumanity. “A powerful indictment of the anti-rationalist attitudes that are dominating our national human race itself. -

Addictions and the Brain
9/18/2012 Addictions and the Brain TAAP Conference September 14, 2012 Acknowledgements • La Hacienda Treatment Center • American Society of Addiction Medicine • National Institute of Drug Abuse © 2012 La Hacienda Treatment Center. All rights reserved. 1 9/18/2012 Definition • A primary, progressive biochemical, psychosocial, genetically transmitted chronic disease of relapse who’s hallmarks are denial, loss of control and unmanageability. DSM IV Criteria for dependency: At least 3 of the 7 below 1. Withdrawal 2. Tolerance 3. The substance is taken in larger amounts or over a longer period than was intended. 4. There is a persistent desire or unsuccessful efforts to cut down or control substance use. 5. A great deal of time is spent in activities necessary to obtain the substance, use the substance, or recover from its effects. 6. Important social, occupational, or recreational activities are given up or reduced because of the substance use. 7. The substance use is continued despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by the substance. © 2012 La Hacienda Treatment Center. All rights reserved. 2 9/18/2012 Dispute between behavior and disease Present understanding of the Hypothalamus location of the disease hypothesis. © 2012 La Hacienda Treatment Center. All rights reserved. 3 9/18/2012 © 2012 La Hacienda Treatment Center. All rights reserved. 4 9/18/2012 © 2012 La Hacienda Treatment Center. All rights reserved. 5 9/18/2012 Dispute regarding behavior versus disease © 2012 La Hacienda Treatment Center. All rights reserved. 6 9/18/2012 © 2012 La Hacienda Treatment Center. -

Antiemetics/Antivertigo Agents
Antiemetic Agents Therapeutic Class Review (TCR) May 1, 2019 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. May 2019 Proprietary Information. Restricted Access – Do not disseminate or copy without approval. © 2004-2019 Magellan Rx Management. All Rights Reserved. 3 FDA-APPROVED INDICATIONS Drug Manufacturer Indication(s) NK1 receptor antagonists aprepitant capsules generic, Merck In combination with other antiemetic agents for: (Emend®)1 . -

Scopolamine As a Potential Treatment Option in Major Depressive Disorder - a Literature Review
Isr J Psychiatry - Vol. 58 - No 1 (2021) Scopolamine as a Potential Treatment Option in Major Depressive Disorder - A Literature Review Dusan Kolar, MD, MSc, PhD, FRCPC Department of Psychiatry, Queen’s University, Mood Disorders Research and Treatment Service, Kingston, Ontario, Canada as selective serotonin reuptake inhibitors (SSRIs) and ABSTRACT serotonin norepinephrine reuptake inhibitors (SNRIs), are currently used as first-line pharmacological treat- Introduction: Slow onset response to antidepressants ments for major depressive disorder (MDD). Despite the and partial response is a common problem in mood availability of a comprehensive list of antidepressants, disorders psychiatry. There is an ongoing need for rapid- approximately 50% of the patients on an antidepressant acting antidepressants, particularly after introducing regimen experience non-response to treatment (3, 4). ketamine in the treatment of depression. Scopolamine Furthermore, the mood elevating effects of antidepres- may have promise as an antidepressant. sant medication is often delayed (5). It is recommended Methods: The author conducted a literature review to that the patient be treated for at least six weeks with identify available treatment trials of scopolamine in the adequate dosage of the given antidepressant before unipolar and bipolar depression in PubMed, the Cochrane considering making changes to the treatment (5). Partial database, Ovid Medline and Google Scholar. to no response to treatment with antidepressants and delayed onset of action indicate that there is a need for Results: There have been eight treatment trials of the development of novel and improved medications scopolamine in MDD and bipolar depression. Seven for the treatment of depression. Many studies in recent studies confirmed significant antidepressant effects years have recognised two different classes of drugs with of scopolamine used as monotherapy and as an rapid and robust antidepressant effects. -

“STOP” and “GO” Pathways for the Treatment of Alcohol Use Disorders
UCSF UC San Francisco Previously Published Works Title Targeting the intracellular signaling "STOP" and "GO" pathways for the treatment of alcohol use disorders. Permalink https://escholarship.org/uc/item/6hd3x2cv Journal Psychopharmacology, 235(6) ISSN 0033-3158 Authors Ron, Dorit Berger, Anthony Publication Date 2018-06-01 DOI 10.1007/s00213-018-4882-z Peer reviewed eScholarship.org Powered by the California Digital Library University of California Psychopharmacology (2018) 235:1727–1743 https://doi.org/10.1007/s00213-018-4882-z REVIEW Targeting the intracellular signaling “STOP” and “GO” pathways for the treatment of alcohol use disorders Dorit Ron1 & Anthony Berger1 Received: 18 January 2018 /Accepted: 12 March 2018 /Published online: 14 April 2018 # The Author(s) 2018 Abstract In recent years, research has identified the molecular and neural substrates underlying the transition of moderate “social” con- sumption of alcohol to the characteristic alcohol use disorder (AUD) phenotypes including excessive and compulsive alcohol use which we define in the review as the GO signaling pathways. In addition, growing evidence points to the existence of molecular mechanisms that keep alcohol consumption in check and that confer resilience for the development of AUD which we define herein as the STOP signaling pathways. In this review, we focus on examples of the GO and the STOP intracellular signaling pathways and discuss our current knowledge of how manipulations of these pathways may be used for the treatment of AUD. Keywords Alcohol . Addiction . Signaling . Translation . Medication Development . Fyn . mTOR . BDNF . GDNF Introduction medications such as naltrexone, acamprosate, and disulfiram not only are beneficial but also suffer from efficacy and com- Alcohol use disorder (AUD) is a serious worldwide health prob- pliance issues (Wackernah et al. -

The Promise of Ketamine
Neuropsychopharmacology (2015) 40, 257–258 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Editorial Circumspectives: The Promise of Ketamine William A Carlezon*,1 and Tony P George2 1 2 Department of Psychiatry, Harvard Medical School, McLean Hospital, Belmont, MA, USA; Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada Neuropsychopharmacology (2015) 40, 257–258; doi:10.1038/npp.2014.270 In this issue we reprise a Feature called Circumspectives. offering hope that fast-acting but safe antidepressants are The general format of a Circumspectives article is similar to possible. a debate, with separate sections in which two thought Despite growing enthusiasm for ketamine and its leaders articulate their individual positions on a topic of promise, Dr Schatzberg describes some sobering details great importance to our community of researchers. The and gaps in knowledge. Ketamine is a drug of abuse and, distinguishing element, however, is that the piece ends with despite some exceptionally elegant studies on the mechan- a ‘reconciliation’ that is co-authored by both and includes ism (eg, Li et al, 2010; Autry et al, 2011), there is no ideas or experiments that will move the field forward. consensus on how it produces therapeutic effects. As one The current Circumspectives (Sanacora and Schatzberg, example, Dr Schatzberg points out similarities in some of 2015) is entitled ‘Ketamine: Promising Path or False the molecular actions of ketamine and scopolamine, Prophecy in the Development of Novel Therapeutics for another familiar and long-standing member of our phama- Mood Disorders?’. It is co-authored by Gerard Sanacora and copea shown to produce rapid antidepressant effects (Furey Alan F Schatzberg, who are leaders in this field. -

Interplay Between Gating and Block of Ligand-Gated Ion Channels
brain sciences Review Interplay between Gating and Block of Ligand-Gated Ion Channels Matthew B. Phillips 1,2, Aparna Nigam 1 and Jon W. Johnson 1,2,* 1 Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA; [email protected] (M.B.P.); [email protected] (A.N.) 2 Center for Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA * Correspondence: [email protected]; Tel.: +1-(412)-624-4295 Received: 27 October 2020; Accepted: 26 November 2020; Published: 1 December 2020 Abstract: Drugs that inhibit ion channel function by binding in the channel and preventing current flow, known as channel blockers, can be used as powerful tools for analysis of channel properties. Channel blockers are used to probe both the sophisticated structure and basic biophysical properties of ion channels. Gating, the mechanism that controls the opening and closing of ion channels, can be profoundly influenced by channel blocking drugs. Channel block and gating are reciprocally connected; gating controls access of channel blockers to their binding sites, and channel-blocking drugs can have profound and diverse effects on the rates of gating transitions and on the stability of channel open and closed states. This review synthesizes knowledge of the inherent intertwining of block and gating of excitatory ligand-gated ion channels, with a focus on the utility of channel blockers as analytic probes of ionotropic glutamate receptor channel function. Keywords: ligand-gated ion channel; channel block; channel gating; nicotinic acetylcholine receptor; ionotropic glutamate receptor; AMPA receptor; kainate receptor; NMDA receptor 1. Introduction Neuronal information processing depends on the distribution and properties of the ion channels found in neuronal membranes. -

From NMDA Receptor Hypofunction to the Dopamine Hypothesis of Schizophrenia J
REVIEW The Neuropsychopharmacology of Phencyclidine: From NMDA Receptor Hypofunction to the Dopamine Hypothesis of Schizophrenia J. David Jentsch, Ph.D., and Robert H. Roth, Ph.D. Administration of noncompetitive NMDA/glutamate effects of these drugs are discussed, especially with regard to receptor antagonists, such as phencyclidine (PCP) and differing profiles following single-dose and long-term ketamine, to humans induces a broad range of exposure. The neurochemical effects of NMDA receptor schizophrenic-like symptomatology, findings that have antagonist administration are argued to support a contributed to a hypoglutamatergic hypothesis of neurobiological hypothesis of schizophrenia, which includes schizophrenia. Moreover, a history of experimental pathophysiology within several neurotransmitter systems, investigations of the effects of these drugs in animals manifested in behavioral pathology. Future directions for suggests that NMDA receptor antagonists may model some the application of NMDA receptor antagonist models of behavioral symptoms of schizophrenia in nonhuman schizophrenia to preclinical and pathophysiological research subjects. In this review, the usefulness of PCP are offered. [Neuropsychopharmacology 20:201–225, administration as a potential animal model of schizophrenia 1999] © 1999 American College of is considered. To support the contention that NMDA Neuropsychopharmacology. Published by Elsevier receptor antagonist administration represents a viable Science Inc. model of schizophrenia, the behavioral and neurobiological KEY WORDS: Ketamine; Phencyclidine; Psychotomimetic; widely from the administration of purportedly psychot- Memory; Catecholamine; Schizophrenia; Prefrontal cortex; omimetic drugs (Snyder 1988; Javitt and Zukin 1991; Cognition; Dopamine; Glutamate Jentsch et al. 1998a), to perinatal insults (Lipska et al. Biological psychiatric research has seen the develop- 1993; El-Khodor and Boksa 1997; Moore and Grace ment of many putative animal models of schizophrenia. -

Auckland Uniservices Limited
Auckland UniServices Limited Legally available, unclassified psychoactive substances and illegal drugs in New Zealand before and after the ban on BZP: a web‐ based survey of patterns of use FINAL REPORT OF FINDINGS June 2009 Janie Sheridan, PhD, BPharm, BA, FRPharmS, RegPharmNZ Rachael Butler, BA, PGDipPH Christine Y. Dong, BSc Hons, BCom Hons Joanne Barnes, PhD, BPharm, MRPharmS, RegPharmNZ, FLS The School of Pharmacy The University of Auckland New Zealand TABLE OF CONTENTS 1 Executive Summary ........................................................................................... 7 2 Introduction .................................................................................................... 11 2.1 Background .............................................................................................. 11 2.1.1 The legislative and regulatory background ................................ 11 2.1.2 The current study ........................................................................ 12 2.2 Study aims ................................................................................................ 12 2.3 Study methods ......................................................................................... 13 2.4 Ethics approval ......................................................................................... 13 2.5 Structure of this report ............................................................................ 13 3 Adverse effects associated with herbal substances used for recreational purposes: a literature review -

Memantine Increases NMDA Receptor Level in the Prefrontal Cortex
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Dundee Online Publications University of Dundee Memantine increases NMDA receptor level in the prefrontal cortex but fails to reverse apomorphine-induced conditioned place preference in rats Ndlazi, Ziphozethu; Abboussi, Oualid; Mabandla, Musa; Daniels, Willie Published in: AIMS Neuroscience DOI: 10.3934/NEUROSCIENCE.2018.4.211 Publication date: 2018 Document Version Publisher's PDF, also known as Version of record Link to publication in Discovery Research Portal Citation for published version (APA): Ndlazi, Z., Abboussi, O., Mabandla, M., & Daniels, W. (2018). Memantine increases NMDA receptor level in the prefrontal cortex but fails to reverse apomorphine-induced conditioned place preference in rats. AIMS Neuroscience, 5(4), 211-220. https://doi.org/10.3934/NEUROSCIENCE.2018.4.211 General rights Copyright and moral rights for the publications made accessible in Discovery Research Portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from Discovery Research Portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim.