Genetic Alteration of the Metal/Redox Modulation of Cav3.2 T-Type
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

7.016 Introductory Biology Fall 2018
7.016: Fall 2018: MIT 7.016 Recitation 15 – Fall 2018 (Note: The recitation summary should NOT be regarded as the substitute for lectures) (This material is COPYRIGHT protected.) Summary of Lecture 22 (11/2): Neurons and action potentials: Ions can move across membranes through pumps and channels. Integral membrane proteins like these cross the membrane via transmembrane domains. Pumps are ATPases that set up the concentration gradients of ions across cell membranes, such that K+ is high inside cells and other ions (such as Cl–, Na+, and Ca2+) are high outside cells. A membrane potential is only set up by ions that move freely across the membrane through open channels, creating one side of the membrane that is more positive relative to the other side. This movement of ions does not dissipate the concentration gradient because the number of ions that move to generate a membrane potential is very small compared to the number of ions that need to be pumped to create a concentration gradient. Most cell membranes only contain open K+ channels and thus only K+ flows freely across membranes through channels; K+ is high inside, causing it to flow outside, giving the inside of cells a negative membrane potential. Ions can move across the membrane through open ion channels. Two forces act to dictate this movement – the concentration gradient and the electrical gradient. Ions move down their concentration gradient through channels, and ions move towards the side of the membrane that harbors the opposite charge. Neurons are the cells of your nervous system that make connections with each other to transmit impulse/ signals. -

Electrical Activity of the Heart: Action Potential, Automaticity, and Conduction 1 & 2 Clive M
Electrical Activity of the Heart: Action Potential, Automaticity, and Conduction 1 & 2 Clive M. Baumgarten, Ph.D. OBJECTIVES: 1. Describe the basic characteristics of cardiac electrical activity and the spread of the action potential through the heart 2. Compare the characteristics of action potentials in different parts of the heart 3. Describe how serum K modulates resting potential 4. Describe the ionic basis for the cardiac action potential and changes in ion currents during each phase of the action potential 5. Identify differences in electrical activity across the tissues of the heart 6. Describe the basis for normal automaticity 7. Describe the basis for excitability 8. Describe the basis for conduction of the cardiac action potential 9. Describe how the responsiveness relationship and the Na+ channel cycle modulate cardiac electrical activity I. BASIC ELECTROPHYSIOLOGIC CHARACTERISTICS OF CARDIAC MUSCLE A. Electrical activity is myogenic, i.e., it originates in the heart. The heart is an electrical syncitium (i.e., behaves as if one cell). The action potential spreads from cell-to-cell initiating contraction. Cardiac electrical activity is modulated by the autonomic nervous system. B. Cardiac cells are electrically coupled by low resistance conducting pathways gap junctions located at the intercalated disc, at the ends of cells, and at nexus, points of side-to-side contact. The low resistance pathways (wide channels) are formed by connexins. Connexins permit the flow of current and the spread of the action potential from cell-to-cell. C. Action potentials are much longer in duration in cardiac muscle (up to 400 msec) than in nerve or skeletal muscle (~5 msec). -

4-Nervous-System-Structure-PPT.Pdf
Nervous Systems: Neuron Structure and Function Integration An animal needs to function like a coherent organism, not like a loose collection of cells. Integration = refers to processes such as summation and coordination that produce coherency and result in harmonious function. Integration Cellular integration = processes within cells Whole-animal integration = selective combination and processing of sensory, endocrine, and central nervous system (CNS) information in ways that promote harmonious functioning of the whole organism within its environment. ◦ This includes its all its cells, tissues, and organs Integration Nerve cells are specialized for control and coordination. Integration ensures that an animal’s responses are smooth and coordinated rather than clashing or disjointed. Excitable Cells Neurons are a type of excitable cell ◦ Specially adapted to generate an electrical signal Can rapidly alter their membrane potential in response to an incoming signal. Vary in structure and function but use the same basic mechanism to send signals. Neuron Function – Main Points Specialized for processing and conveying information. Information is coded into changes in electrical potential across cell membranes. Neurons use action potentials to transmit these signals across long distances. Neurons allow animals to sense and respond to their environment. Benefits of Neurons Plants (no neurons): Action potentials travel @ 1-3 cm/sec Animals (neurons): Action potentials travel @ 100m/sec or 10,000cm/sec CNS to Muscles Signal Reception Dendrites & Cell Body Signal Integration Axon Hillock Signal Conduction Axon Signal Transmission Axon Terminals Signal Reception Dendrites sense and convert incoming signals into electrical signals by changing membrane potential. Cell Body = routine metabolic functions Signal Integration Incoming signals are conducted to the axon hillock If signal is sufficiently large an electrical signal, or action potential, is initiated. -

Electrolyte Disorders and Arrhythmogenesis
Cardiology Journal 2011, Vol. 18, No. 3, pp. 233–245 Copyright © 2011 Via Medica REVIEW ARTICLE ISSN 1897–5593 Electrolyte disorders and arrhythmogenesis Nabil El-Sherif1, Gioia Turitto2 1State University of NY, Downstate Medical Center and NY Harbor VA Healthcare System, Brooklyn, NY, USA 2Methodist University Hospital, Brooklyn, NY, USA Abstract Electrolyte disorders can alter cardiac ionic currents kinetics and depending on the changes can promote proarrhythmic or antiarrhythmic effects. The present report reviews the mecha- nisms, electrophysiolgical (EP), electrocardiographic (ECG), and clinical consequences of elec- trolyte disorders. Potassium (K+) is the most abundent intracellular cation and hypokalemia is the most commont electrolyte abnormality encountered in clinical practice. The most signifcant ECG manifestation of hypokalemia is a prominent U wave. Several cardiac and + non cardiac drugs are known to suppress the HERG K channel and hence the IK, and especially in the presence of hypokalemia, can result in prolonged action potential duration and QT interval, QTU alternans, early afterdepolarizations, and torsade de pointes ventricu- lar tachyarrythmia (TdP VT). Hyperkalemia affects up to 8% of hospitalized patients mainly in the setting of compromised renal function. The ECG manifestation of hyperkalemia de- pends on serum K+ level. At 5.5–7.0 mmol/L K+, tall peaked, narrow-based T waves are seen. At > 10.0 mmol/L K+, sinus arrest, marked intraventricular conduction delay, ventricular techycardia, and ventricular fibrillation can develop. Isolated abnormalities of extracellular calcium (Ca++) produce clinically significant EP effects only when they are extreme in either direction. Hypocalcemia, frequently seen in the setting of chronic renal insufficiency, results in prolonged ST segment and QT interval while hypercalcemia, usually seen with hyperparathy- roidism, results in shortening of both intervals. -

Two Components of the Cardiac Action Potential I
Two Components of the Cardiac Action Potential I. Voltage-time course and the effect of acet.flcholine on atrial and nodal cells of the rabbit heart ANTONIO PAES de CARVALHO, BRIAN FRANCIS HOFFMAN, and MARILENE de PAULA CARVALHO From the Instituto de Biofisica da Universidade Federal do Rio de Janeiro, Rio de Janeiro (GB), Brazil, and the Department of Pharmacology, College of Physicians and Surgeons, Columbia University, New York 10032 ABSTRACT Transmembrane potentials recorded from the rabbit heart in vitro were displayed as voltage against time (V, t display), and dV/dt against voltage (I?, V or phase-plane display). Acetylcholine was applied to the record- ing site by means of a hydraulic system. Results showed that (a) differences in time course of action potential upstroke can be explained in terms of the rela- tive magnitude of fast and slow phases of depolarization; (b) acetylcholine is capable of depressing the slow phase of depolarization as well as the plateau of the action potential; and (¢) action potentials from nodal (SA and AV) cells seem to lack the initial fast phase. These results were construed to support a two-component hypothesis for cardiac electrogenesis. The hypothesis states that cardiac action potentials are composed of two distinct and physiologically separable "components" which result from discrete mechanisms. An initial fast component is a sodium spike similar to that of squid nerve. The slow com- ponent, which accounts for both a slow depolarization during phase 0 and the plateau, probably is dependent on the properties of a slow inward current hav- ing a positive equilibrium potential, coupled to a decrease in the resting potas- sium conductance. -

V=Nx76xs 4FRE the Following Content Is Provided Under a Creative Commons License
MITOCW | watch?v=Nx76XS_4FRE The following content is provided under a Creative Commons license. Your support will help MIT OpenCourseWare continue to offer high quality educational resources for free. To make a donation or view additional materials from hundreds of MIT courses, visit MIT OpenCourseWare at ocw.mit.edu. HAZEL SIVE: Some good questions. Let's zip through quickly. Why can't an initial depolarization travel along the length of the axon? Last time, we went through in detail how a signal is transmitted from the outside to the inside of the cell by ion flux across the membrane, and then how that ion flux is transmitted down the neuron. Remember that the ions entering the cell are just ions. They'll diffuse away. And so the depolarization kind of dissipates. And it then needs to be regenerated. The action potential, each small depolarization or the whole signal, each small depolarization with a specific magnitude, the reversal of membrane potential going from about minus 60 millivolts, negative inside, to plus 55 or 60 millivolts, positive inside. Each of those is an action potential. And here is an interesting one. Why doesn't the membrane potential stop at zero when sodium rushes into the cell during an action potential, which is what you'd expect if sodium was just moving along its concentration radian? Because the potential's not only governed by sodium influx. There are chloride ions outside the cell and potassium ions inside over the duration of the potential. And as we'll discuss briefly in a moment, it's during the rectification process that you go back to zero, and then you reverse membrane potential again. -

Electrophysiological Foundations of Cardiac Arrhythmias Clinical Electrophysiology Mark E
Second Edition Electrophysiological Foundations Andrew L. Wit of Cardiac Arrhythmias Penelope A. Boyden A Bridge Between Basic Mechanisms and Electrophysiological Foundations of Cardiac Arrhythmias Electrophysiological Foundations of Cardiac Clinical Electrophysiology Mark E. Josephson Second Edition Hein J. Wellens Now in an abridged second edition, Electrophysiological Foundations of Cardiac Arrhythmias focuses on teaching the fundamental concepts of cardiac cellular • Boyden Josephson Wellens Wit electrophysiology with an emphasis on the relationship of basic mechanisms to clinical cardiac arrhythmias. Understanding this relationship and the electrophysiological mechanisms underlying arrhythmogenesis will be invaluable Electrophysiological to physicians entering the fields of cardiology and clinical electrophysiology, as well as those scientists and clinicians already working in these areas. These essential concepts of electrophysiology include discussion on action potentials, ion channels and currents, and mechanisms of arrhythmias, and Foundations of provide the working knowledge that will enable the reader to approach a board exam confidently. Additionally, the authors build a base of understanding that will prepare the reader for more advanced texts, such as Josephson’s Clinical Cardiac Electrophysiology: Techniques and Interpretations. Cardiac Arrhythmias About the Authors The authors, Andrew L. Wit, PhD, Penelope A. Boyden, PhD, Mark E. Josephson, A Bridge Between MD, and Hein J. Wellens, MD, PhD, have spent four lifetimes investigating and defining electrophysiological mechanisms of cardiac arrhythmias. But equally important, they have devoted their careers to teaching this subject to students of all levels, from medical Basic Mechanisms and students to more advanced cardiology and clinical electrophysiology fellows. Clinical Electrophysiology ISBN 978-1-942909-42-2 Second Edition 90000> This book includes access to a free digital edition for use by the first buyer. -

NEURON and the PHYSIOLOGY of NERVE IMPULSE the Nervous
NEURON AND THE PHYSIOLOGY OF NERVE IMPULSE The nervous system is made of millions of structural and functional units called nerve cell or neurons and a number of supporting cells called glial cells. The function of the nerve cell is to receive and transmit impulses or messages and integrate the entire body as a single unit. The neurons can be classified into three types – 1. Unipolar neuron- it has a cell body or cyton from which arises a single short branch called neurite that immediately divides into the dendritic or receiving branch and axonic or transmitting branch. 2. Bipolar neuron- The neuron has the cell body from which arises two neurites called the dendron and the axon respectively. 3. Multipolar neuron- From the cell body arise many dendrites and one long axon. Neurons can also be classified into 1) myelinated when myelin sheath surrounds the axon and 2) non-myelinated neurons when the axon is naked and not covered by the myelin sheath. 1 Structure of a myelinated neuron/nerve cell The neuron can be divided into three parts-cell body or cyton from which arises two types of branches, i.e., many branched processes called dendrons and a long unbranched process called axon. The cyton or cell body is filled with the cytoplasm also called neuroplasm that is limited by the plasma membrane. It contains the nucleus, mitochondria, endoplasmic reticulum, golgi bodies, neurofibrils (filaments that act as a cytoskeleton) and special dense RNA rich granules called Nissl’s granules (help in protein synthesis). The dendrons terminate in slender processes called dendrites. -
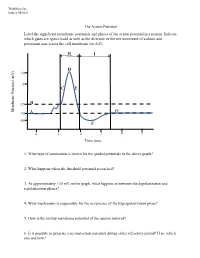
The Action Potential Label the Significant Membrane Potentials and Phases of the Action Potential in a Neuron
Worksheet by Andrey Michel The Action Potential Label the significant membrane potentials and phases of the action potential in a neuron. Indicate which gates are open/closed as well as the direction of the net movement of sodium and potassium ions across the cell membrane for A-G. H I D +30 0 C E -55 B Membrane Potential Membrane (mV) G -70 A -80 F 0 1 2 3 4 5 Time (ms) 1. What type of summation is shown for the graded potentials in the above graph? 2. What happens when the threshold potential is reached? 3. At approximately +30 mV on the graph, what happens in between the depolarization and repolarization phases? 4. What mechanism is responsible for the occurrence of the hyperpolarization phase? 5. How is the resting membrane potential of the neuron restored? 6. Is it possible to generate a second action potential during either refractory period? If so, which one and how? Place the following events in chronological order from 1-8: Na+ enters the cell, and depolarization occurs to approximately +30 mV. The voltage across the cell membrane is -70 mV, the resting membrane potential. Upon reaching the peak of the action potential, the VG Na+ channels are inactivated by the closing of their inactivation gate and the activation gate of each VG K+ channel opens. VG K+ channels close by the closing of their activation gate, and the resting membrane potential is gradually restored. An excitatory post-synaptic potential depolarizes the membrane to threshold and the activation gate of VG Na+ channels open. -

The Voltage-Gated Channels of Ca2 Action Potentials: Generalization
CHAPTER 5 The voltage-gated channels of Ca2ϩ action potentials: Generalization Chapter 4 explained the Naϩ-dependent action will examine the properties of a Ca2ϩ-dependent potential propagated by axons. There are two other action potential. types of action potentials: (i) the Naϩ/Ca2ϩ-dependent action potential present in axon terminals or heart 2؉ ؉ muscle cells (Figure 4.2d), for example, where it is 5.1.1 Ca and K ions participate in the responsible for Ca2ϩ entry and an increase of intracel- action potential of endocrine cells lular Ca2ϩ concentration, a necessary prerequisite for The activity of pituitary endocrine cells that release neurotransmitter release (secretion) or muscle fibre growth hormone is recorded in the perforated whole- contraction; and (ii) the Ca2ϩ-dependent action poten- cell configuration (current clamp mode; see Appendix tial (in which Naϩ ions do not participate) in dendrites 4.1). They display a spontaneous activity. When these of Purkinje cells of the cerebellum (see Figure 17.9) and cells are previously loaded with the Ca2ϩ-sensitive dye in endocrine cells (Figure 5.1a). In Purkinje cell den- Fura-2, changes of intracellular Ca2ϩ concentration can drites, it depolarizes the membrane and thus modu- be also quantified (see Appendix 5.1). Simultaneous 2ϩ lates neuronal integration; in endocrine cells it recording of potential and [Ca ]i changes shows that 2ϩ 2ϩ provides a Ca entry to trigger hormone secretion. for each action potential there is a corresponding [Ca ]i increase (Figure 5.1a). This strongly suggests -

Biology 251 Fall 2015 1 TOPIC 4: ACTION POTENTIALS AND
Biology 251 Fall 2015 TOPIC 4: ACTION POTENTIALS AND GRADED POTENTIALS I. Introduction A. All cells possess a membrane potential related to the non-uniform distribution of, and differential permeability of, Na+, K+ and A-. B. Nerve and muscle are excitable tissue that use this potential by undergoing controlled, transient, rapid changes in membrane potential. Such fluctuations in membrane potential serve as electrical signals. C. Two kinds of such electrical signals 1. Graded potentials are short distance signals 2. Action potentials are long distance signals II. Action Potentials A. Terms (Fig 7.11) 1. Polarization: separation of charges 2. Depolarization: reduction in potential 3. Hyperpolarization: increase in potential 4. Repolarization: return to resting potential B. Events in an Action Potential (Fig 7.16) 1. Occurrence of a triggering event 2. Slow depolarization from -70 mV to -55 mV 3. -55 mV is threshold potential; as soon as it is reached, a very rapid depolarization to +30 mV occurs 4. repolarization then occurs 5. frequently overshoots to -80 mV before repolarizing at -70 mV; this is hyperpolarization 6. in a nerve, this whole process takes 0.001 seconds C. Mechanisms underlying events in Action Potential (Fig 7.16, 7.17) 1. Membrane contains Na+ channels and K+ channels. Both of these are voltage regulated channels, meaning that changes in voltage (i.e., membrane potential) open or close them. 2. At rest (-70 mV) some K+ channels are open, fewer Na+ channels open; the “leaking” of these two ions across membrane balanced by Na-K-ATPase pump (see Topic 3-VI-C-4) 3. -

BRS Physiology Cases and Problems 2Nd Edition
CELLULAR AND AUTONOMIC PHYSIOLOGY 19 Case 4 Primary Hypokalemic Periodic Paralysis Jimmy Jaworski is a 16-year-old sprinter on the high school track team. Recently, after he com- pleted his events, he felt extremely weak, and his legs became "like rubber." Eating, especially carbohydrates, made him feel worse. After the most recent meet, he was unable to walk and had to be carried from the track on a stretcher. His parents were very alarmed and made an appoint- ment for Jimmy to be evaluated by his pediatrician. As part of the workup, the pediatrician mea- sured Jimmy's serum K. concentration, which was normal (4.5 rnEq/L). However, because the pediatrician suspected a connection with K*, the measurement was repeated immediately after a strenuous exercise treadmill test. After the treadmill test, Jimmy's serum K- was alarmingly low (2.2 mEq/L). Jimmy was diagnosed as having an inherited disorder called primary hypokalemic periodic paralysis and subsequently was treated with I(' supplementation. QUESTIONS 1. What is the normal distribution of I(' between intracellular fluid and extracellular fluid? Where is most of the K. located? 2. What major factors can alter the distribution of K . between intracellular fluid and extracellular fluid? 3. What is the relationship between the serum K. concentration and the resting membrane poten- tial of excitable cells (e.g., nerve, skeletal muscle)? 4. How does a decrease in serum K+ concentration alter the resting membrane potential of skeletal muscle? 5. Propose a mechanism whereby a decrease in the serum K . concentration could lead to skeletal muscle weakness.