Molecular Biology in Cellular Pathology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chlamydia Trachomatis Infection Is Driven by Nonprotective Immune Cells That Are Distinct from Protective Populations
Pathology after Chlamydia trachomatis infection is driven by nonprotective immune cells that are distinct from protective populations Rebeccah S. Lijeka,b,1, Jennifer D. Helblea, Andrew J. Olivea,c, Kyra W. Seigerb, and Michael N. Starnbacha,1 aDepartment of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115; bDepartment of Biological Sciences, Mount Holyoke College, South Hadley, MA 01075; and cDepartment of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Rafi Ahmed, Emory University, Atlanta, GA, and approved December 27, 2017 (received for review June 23, 2017) Infection with Chlamydia trachomatis drives severe mucosal immu- sequence identity, Chlamydia muridarum, the extent to which the nopathology; however, the immune responses that are required for molecular pathogenesis of C. muridarum represents that of Ct is mediating pathology vs. protection are not well understood. Here, unknown (6). Ct serovar L2 (Ct L2) is capable of infecting the we employed a mouse model to identify immune responses re- mouse upper genital tract when inoculated across the cervix into quired for C. trachomatis-induced upper genital tract pathology the uterus (7, 8) but it does not induce robust immunopathology. and to determine whether these responses are also required for This is consistent with the human disease phenotype caused by Ct L2, bacterial clearance. In mice as in humans, immunopathology was which disseminates to the lymph nodes causing lymphogranuloma characterized by extravasation of leukocytes into the upper genital venereum (LGV) and is not a major cause of mucosal immunopa- thology in the female upper genital tract (uterus and ovaries). tract that occluded luminal spaces in the uterus and ovaries. -

Department of Experimental Pathology, Immunology and Microbiology 531
Department of Experimental Pathology, Immunology and Microbiology 531 Department of Experimental Pathology, Immunology and Microbiology Interim Chairperson: Zaatari, Ghazi Vice Chairperson: Matar, Ghassan Professors: Abdelnoor, Alexander; Khouri, Samia; Matar, Ghassan; Sayegh, Mohamed; Zaatari, Ghazi Associate Professor: Rahal, Elias Assistant Professors: Al-Awar, Ghassan; El Hajj, Hiba; Shirinian, Margret; Zaraket, Hassan The Department of Experimental Pathology, Immunology and Microbiology offers courses to medical laboratory sciences (MLSP) students as well as nursing, medical, and graduate students. It offers a graduate program (discipline of Microbiology and Immunology) leading to a master’s degree (MS) or doctoral degree (PhD) in Biomedical sciences. The requirements for admission to the graduate program are stated on page 33 of this catalogue. IDTH 203 The immune System in Health and Disease 37.28; 3 cr. See Interdepartmental Courses. IDTH 205 Microbiology and Infectious Diseases 37.28; 5 cr. See Interdepartmental Courses. MBIM 223 Parasitology for MLSP Students 39.39; 4 cr. Second semester. MBIM 237 Microbiology and Immunology for Nursing Degree Students 32.64; 3 cr. A course on the fundamental aspects of medical microbiology and immunology for nursing students. Second semester. MBIM 260 Elective in Infectious Diseases for Medicine III and IV 0.180 A course on basic evaluation, diagnosis, and management of infectious diseases. One month. MBIM 261 Elective in Immunology for Medicine III and IV 0.180 A course that is an introduction to immunological research and its application to clinical practice. One month. MBIM 310 Basic Immunology 32.32; 3 cr. A course on innate and adaptive immune mechanisms, infection and immunity, vaccination, immune mechanisms in tissue injury and therapeutic immunology. -

Deciphering the Triad of Infection, Immunity and Pathology
INSIGHT DISEASE Deciphering the triad of infection, immunity and pathology The factors which drive and control disease progression can be inferred from mathematical models that integrate measures of immune responses, data from tissue sampling and markers of infection dynamics. FREDERIK GRAW immune actors in the body. Now, in eLife, Related research article Myers MA, Smith Amber Smith and colleagues at St. Jude Child- AP, Lane LC, Moquin DJ, Aogo R, Woolard ren’s Research Hospital, the University of Ten- S, Thomas P, Vogel P, Smith AM. 2021. nessee Health Science Center and the Dynamically linking influenza virus infection Washington University School of Medicine – kinetics, lung injury, inflammation, and dis- including Margaret Myers and Amanda Smith as ease severity. eLife 10:e68864. doi: 10. joing first authors – report how viral infection, 7554/eLife.68864 counteracting immune responses and lung pathology interact as mice fight off influenza A (Myers et al., 2021). First, the team tracked how viral load and the number of CD8+ T cells, an important immune fever, a cough, a splitting headache... actor that helps to clear infected cells, pro- Being sick often comes with tell-tale gressed over time. In combination with mathe- A signs which worsen as the disease pro- matical models, these measurements allowed gresses and tissues become damaged. These Myers et al. to estimate several parameters that symptoms result from complex interactions reflect the pace at which the virus replicates, the between the infecting pathogen, the inflamma- strength of the immune response, and the inter- tion process, and the response from the immune actions between these processes. -

Overview of Pathology and Its Related Disciplines - Soheir Mahmoud Mahfouz
MEDICAL SCIENCES – Vol.I -Overview of Pathology and its Related Disciplines - Soheir Mahmoud Mahfouz OVERVIEW OF PATHOLOGY AND ITS RELATED DISCIPLINES Soheir Mahmoud Mahfouz Cairo University, Kasr El Ainy Hospital, Egypt Keywords: Pathology, Pathology disciplines, Pathology techniques, Ancillary diagnostic methods, General Pathology, Special Pathology Contents 1. Introduction 1.1 Pathology coverage 1.1.1 Etiology and Pathogenesis of a Disease 1.1.2 Manifestations of Disease (Lesions) 1.1.3 Phases Of A Disease Process (Course) 1.2 Physician’s approach to patient 1.3 Types of pathologists and affiliated specialties 1.4 Role of pathologist 2. Pathology and its related disciplines 2.1 Cytology 2.1.1 Cytology Samples 2.1.2 Technical Aspects 2.1.3 Examination of Sample and Diagnosis 3. Pathology techniques and ancillary diagnostic methods 3.1 Macroscopic pathology 3.2 Light Microscopy 3.3 Polarizing light microscopy 3.4 Electron microscopy (EM) 3.5 Confocal Microscopy 3.6 Frozen section 3.7 Cyto/histochemistry 3.8 Immunocyto/histochemical methods 3.9 Molecular and genetic methods of diagnosis 3.10 Quantitative methods 4. Types of tests used in pathology 4.1 DiagnosticUNESCO tests – EOLSS 4.2 Quantitative tests 4.3 Prognostic tests 5. The scope of SAMPLEpathology & its main divisions CHAPTERS 6. Conclusions Glossary Bibliography Biographical sketch Summary Pathology is the science of disease. It deals with deviations from normal body function and ©Encyclopedia of Life Support Systems (EOLSS) MEDICAL SCIENCES – Vol.I -Overview of Pathology and its Related Disciplines - Soheir Mahmoud Mahfouz structure. Many disciplines are involved in the study of disease, as it is necessary to understand the complex causes and effects of various disorders that affect the organs and body as a whole. -
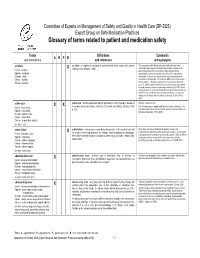
Glossary of Terms Related to Patient and Medication Safety
Committee of Experts on Management of Safety and Quality in Health Care (SP-SQS) Expert Group on Safe Medication Practices Glossary of terms related to patient and medication safety Terms Definitions Comments A R P B and translations and references and synonyms accident accident : an unplanned, unexpected, and undesired event, usually with adverse “For many years safety officials and public health authorities have Xconsequences (Senders, 1994). discouraged use of the word "accident" when it refers to injuries or the French : accident events that produce them. An accident is often understood to be Spanish : accidente unpredictable -a chance occurrence or an "act of God"- and therefore German : Unfall unavoidable. However, most injuries and their precipitating events are Italiano : incidente predictable and preventable. That is why the BMJ has decided to ban the Slovene : nesreča word accident. (…) Purging a common term from our lexicon will not be easy. "Accident" remains entrenched in lay and medical discourse and will no doubt continue to appear in manuscripts submitted to the BMJ. We are asking our editors to be vigilant in detecting and rejecting inappropriate use of the "A" word, and we trust that our readers will keep us on our toes by alerting us to instances when "accidents" slip through.” (Davis & Pless, 2001) active error X X active error : an error associated with the performance of the ‘front-line’ operator of Synonym : sharp-end error French : erreur active a complex system and whose effects are felt almost immediately. (Reason, 1990, This definition has been slightly modified by the Institute of Medicine : “an p.173) error that occurs at the level of the frontline operator and whose effects are Spanish : error activo felt almost immediately.” (Kohn, 2000) German : aktiver Fehler Italiano : errore attivo Slovene : neposredna napaka see also : error active failure active failures : actions or processes during the provision of direct patient care that Since failure is a term not defined in the glossary, its use is not X recommended. -
Pathology: a Career in Medicine the Study of the Nature of Disease, Its Causes, Processes, Development, and Consequences
PATHOLOGY A Career in Medicine The Intersociety Council for Pathology Information (ICPI) www.pathologytraining.org 2015 Pathology: A Career in Medicine The study of the nature of disease, its causes, processes, development, and consequences. Pathology is the medical specialty that provides a scientific foundation for medical practice The pathologist is a physician who specializes in the diagnosis and management of human disease by laboratory methods. Pathologists function in three broad areas: as diagnosticians, as teachers, and as investigators. Fundamental to the discipline of pathology is the need to integrate clinical information with physiological, biochemical and molecular laboratory studies, together with observations of tissue alterations. Pathologists in hospital and clinical laboratories practice as consultant physicians, developing and applying knowledge of tissue and laboratory analyses to assist in the diagnosis and treatment of individual patients. As teachers, they impart this knowledge of disease to their medical colleagues, to medical students, and to trainees at all levels. As scientists, they use the tools of laboratory science in clinical studies, disease models, and other experimental systems, to advance the understanding and treatment of disease. Pathology has a special appeal to those who enjoy solving disease-related problems, using technologies based upon fundamental sciences ranging from biophysics to molecular genetics, as well as tools from the more traditional disciplines of anatomy, biochemistry, pharmacology, physiology and microbiology. The Pathologist in Patient Care The pathologist uses diagnostic and screening tests to identify and interpret the changes that characterize different diseases in the cells, tissues, and fluids of the body. Anatomic pathology involves the analysis of the A biosample robot prepares specimens for gross and microscopic structural changes caused by testing disease in tissues and cells removed during biopsy procedures, in surgery, or at autopsy. -

Cytopathology (Path Cyto)
path cyto 1 Pathology: Cytopathology Page updated: August 2020 This section contains information to assist providers in billing for pathology procedures related to cytopathology services. Pap Smear Tests Taking a Papanicolaou (Pap) smear sample is considered part of a pelvic examination and is not separately reimbursable. The Pap smear test is reimbursable only to the provider who performs and reads the Pap smear and issues the written report. These tests include up to three smears for Pap tests for cancer screening and/or a qualitative report on the patient’s level of estrogen. Modifiers TC and 26 Providers may use modifier TC to bill cervical or vaginal Pap smear results. When a smear is billed with modifier 26, it is reimbursable only to a hospital pathologist whose service is not covered by the hospital. Submitting a claim on one line without a modifier indicates that both the professional and technical components of the service were provided. Clinical or Hospital Laboratory Pap smears examined in a clinical or hospital laboratory can be reimbursed without a modifier only if both the professional and technical components are performed in the laboratory. Billing Restrictions CPT® code 88120 will not be reimbursed if billed in conjunction with codes 88121 or 88365; CPT code 88121 will not be reimbursed if billed in conjunction with codes 88120 or 88365. CPT codes 88142, 88143, 88147, 88148, 88150, 88152, 88153, 88164, 88165, 88166 or 88167, 88174 or 88175 may be used to bill cervical or vaginal Pap smear tests and to report physician interpretation services. Part 2 – Cytopathology path cyto 2 Page updated: August 2020 Recommended frequencies are as follows: ‹‹Recommended Frequencies CPT Codes›› CPT Codes Recommended Frequency 88142, 88143, 88147, 88148, 88150, One in 30 days 88152, 88153, 88174, 88175 88164 thru 88167 One in one year Frequency limits apply to claims billed by any provider, for the same recipient. -

General Pathology and Immunology Morton H
General Pathology and Immunology Morton H. Levitt, M.D., Course Director BMS 6601 [email protected] Phone: 644-0498 Fall 2004 Course Syllabus Syllabus The “Course Documents” section in Blackboard includes the PowerPoint lectures for the course. In addition, there will be handouts for all the lectures and group sessions. These are designed to supplement and organize the material in the textbook, but not be a complete substitute for it. Course Objectives The general pathology course in the Fall semester at FSUCOM covers the basic pathophysiology of mechanisms of disease in medicine. The knowledge gained from study of these basic mechanisms will be applied to systemic pathology in the Spring semester and to clerkships in the 3rd and 4th years. This pathology course will incorporate gross pathologic, microscopic, and radiologic material to assist you in understanding the disease processes and prepare you for licensing examinations. The knowledge gained from a study of pathology will integrate with other courses to provide you with the means for assessment and diagnosis of patients under your care. Lectures / Discussions / Tutorials Check locations for lectures in your FSUCOM master schedule. See the course schedule for dates and times. The lectures are designed to cover the course content in an organized fashion, illustrating the concepts and allowing time for you to ask questions. There are 39 lecture hours and 4 discussion/tutorial hours. Laboratories / Small Groups/Problem-Based Learning (PBL) Exercises Check the schedule for times and locations. You will be assigned to one of the laboratory or small group rooms. Assignments for small group sessions will be made on the basis of the number of groups and room availability. -

Pathology (PATH) 1
Pathology (PATH) 1 PATH 599. Ph.D. Thesis Research. 0-16 hours. Pathology (PATH) Research in experimental pathology towards a Ph.D. degree. Course Information: Satisfactory/Unsatisfactory grading only. May be repeated. Courses Students may register in more than one section per term. PATH 421. General Pathology - Dental. 3 hours. Basic principles of pathological processes. Course Information: Prerequisite(s): ANAT 440 and PATH 407 and PHYB 401; or consent of instructor. Class Schedule Information: To be properly registered, students must enroll in one Laboratory and one Lecture. PATH 422. Systemic Pathology - Dentistry. 3 hours. Disease process affecting specific organs. Course Information: Prerequisite(s): PATH 421. Class Schedule Information: To be properly registered, students must enroll in one Laboratory and one Lecture. PATH 507. Physiological Basis of Pathology. 2 hours. Subject matter allied to general pathology but going deeper into physical chemistry and physiological principles, as set forth in N.R. Joseph's "Comparative Physical Biology." Course Information: Same as HSTL 507. Prerequisite(s): HSTL 401; or PATH 421 and PATH 422. PATH 510. General Pathology. 3 hours. Introduction to the science of disease for graduate students in areas of biomedical research with a focus on the mechanisms of basic pathological processes at the molecular, cellular, tissue, and whole organism levels. Course Information: Prerequisite(s): Consent of the instructor. Recommended background: Basic molecular and cell biology is highly recommended. PATH 511. Pathobiology of Cancer. 3 hours. Introduction to principles of carcinogenesis, tumor biology, and oncology, including cancer epidemiology, molecular-cellular basis of cancer, tumor progression, invasion and metastasis, and prevention, detection, diagnosis, and therapy of cancer. -

Course Syllabus
Course Syllabus WELCOME TO THE PATHOLOGY COURSE I apologize for the length of this introductory document. However, it includes the answers to most of the questions raised (often countless times) during previous years. When you have questions, please refer to this document. There's a good chance you will find the answer. If not… next year it'll be even longer! Content is as follows: Contact Information Course Philosophy and Goals Course Objectives Course Materials Policies Professional Expectations Course Updates Based on Student Feedback Concluding Notes CONTACT INFORMATION Coursemaster: Erika Crouch, M.D., Ph.D. Office: BJC Institute of Health Email: [email protected] Phone: (314) 454-8462 Course Coordinator Our Course Coordinator is Ashley Edwards (314-454-8462, [email protected]). Although I am always available to you, it would be helpful if you could initially direct administrative questions to Ashley, most reliably via email. Section and Theme Leaders Each 2nd year block contains one or more sections, each with its own Section Leader. The Course also includes two themes with content that spans two or more blocks. As discussed elsewhere, the organization of sections and themes has changed since last year, primarily to provide more time for end of the year board preparation. This included the conversion of the short Endocrine Pathology section to a "theme" and the move of the OB- GYN/Testicular Pathology from Block 6 to Block 3. The Section or Theme Leaders are responsible for the organization and content of the individual sections or themes, respectively. I'm always available to answer questions. -

Cytopathology Supplemental Guide
Cytopathology Supplemental Guide Supplemental Guide: Cytopathology November 2020 1 Cytopathology Supplemental Guide TABLE OF CONTENTS INTRODUCTION ............................................................................................................................. 3 PATIENT CARE .............................................................................................................................. 4 REPORTING .................................................................................................................................... 4 CONSULTATION .............................................................................................................................. 6 PERFORMANCE OF FINE NEEDLE ASPIRATIONS ................................................................................ 8 ADEQUACY (RAPID ON-SITE EVALUATION [ROSE]) AND TRIAGE ..................................................... 10 FINE NEEDLE ASPIRATION SLIDE AND CORE BIOPSY TOUCH PREPARATIONS ................................... 12 CYTOPREPATORY TECHNIQUES ..................................................................................................... 13 MEDICAL KNOWLEDGE .............................................................................................................. 14 DIAGNOSIS ................................................................................................................................... 14 CLINICAL REASONING .................................................................................................................. -

Pathology (Path) 1
PATHOLOGY (PATH) 1 PATH 715. Molecular and Cellular Pathophysiological Basis of Disease: PATHOLOGY (PATH) Systemic Pathology. 3 Credits. A graduate-level laboratory course on systemic pathology, emphasizing PATH 128. Biology of Human Disease. 3 Credits. diseases of major organ systems. A follow-up to PATH 713/714L. Three Open to all undergraduates. An overview of basic human molecular and lecture hours (three credits) with a complementary two-and-a-half-hour cellular biology in the setting of common human diseases. The course laboratory ( two credits) each week. emphasizes how an understanding of disease mechanisms provides the Requisites: Co-requisite, PATH 716L. knowledge base for informed use of modern health care. Does not count Grading status: Letter grade. as a course in the major. PATH 716L. Molecular and Cellular Pathophysiological Basis of Disease: Grading status: Letter grade. Laboratory II. 2 Credits. PATH 426. Biology of Blood Diseases. 3 Credits. A graduate-level laboratory course on mechanisms of systemic disease An introduction to the biology and pathophysiology of blood and the pathogenesis, emphasizing cell and tissue-based examples of diseases molecular mechanisms of some human diseases: anemias; leukemias; of the major organ systems. hemorrhagic, thrombotic, and vascular disorders; and HIV disease/AIDS. Requisites: Pre- or corequisite, PATH 715. Honors version available Grading status: Letter grade. Requisites: Prerequisite, BIOL 205; Permission of the instructor for PATH 723. Practical Considerations for Translational Research. 2 Credits. students lacking the prerequisite. Permission of the instructor. A multi-disciplinary course providing Grading status: Letter grade students principles involved in translating basic science into clinically Same as: BIOL 426.