Cis Peptidyl Prolyl Helicobacter Pylori Central
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
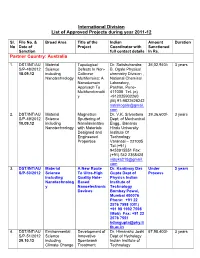
International Division List of Approved Projects During Year 2011-12
International Division List of Approved Projects during year 2011-12 Sl. File No. & Broad Area Title of the Indian Amount Duration No Date of Project Coordinator with Sanctioned Sanction full contact details In Rs. Partner Country: Australia 1. DST/INT/A U Material Topological Dr. Satishchandra 36,02,940/- 3 years S/P-48/2012 Science Defects In Non- B. Ogale Physical 18.09.12 including Collinear chemistry Division , Nanotechnology Multiferroics: A National Chemical Nanodomain Laboratory, Approach To Pashan, Pune- Multifunctionalit 411008 Tel. (o) y +912025902260 (M) 91-9822628242 satishogale@gmail. com 2. DST/INT/AU Material Magnetron Dr. V.K. Srivastava 39,36,600/- 3 years S/P-49/2012 Science Sputtering of Dept. of Mechanical 10.09.12 including Nanolaminates Engg., Banaras Nanotechnology with Materials Hindu University Designed and Institute Of Engineered Technology Properties Varanasi – 221005 Tel.(+91) 9453915551 Fax: (+91) 542 2368428 vijayks210@gmail. com 3. DST/INT/AU Material A New Route Dr. Kantimay Das Under 3 years S/P-50/2012 Science To Ultra-High Gupta Dept of Process including Quality Hole- Physics Indian Nanotechnolog Based Institute of y Nanoelectronic Technology Devices Bombay Powai, Mumbai 400076 Phone: +91 22 2576 7598 (Off.) +91 98 1992 7598 (Mob) Fax: +91 22 2576 7551 [email protected] tb.ac.in 4. DST/INT/AU Environmental Development of Dr. Himanshu Joshi 67,98,400/- 3 years S/P-51/2012 Science Innovative Dept of Hydrology 29.10.12 including Spentwash Indian Institute of Climate Change Treatment Technology Research Systems and Roorkee– 247667, Resource Uttarakhand Recovery himanshujoshi58@ Options for gmail.com; Distillery himanshu_joshi58 Application @yahoo.co.in; [email protected] n 5. -

Centenary Celebration of Bose Institute
C E N T E N A R Y C E L E B R AT I O N CCEENNTTEENNAARRYY CCEELLEEBBRRAATTIIOONN OOFF BBOOSSEE IINNSSTTIITTUUTTEE 1 9 1 7 - 2 0 1 7 List of Posters (24.11.2017–28.11.2017) Poster Faculty Programme Title Poster Faculty Programme Title No No A-1 Amita Pal I Molecular characterization of VmMAPK1 and deciphering its role in restricting D-10 Tanya Das IV Is cancer a stem cell disease? MYMIV multiplication in tobacco Poulami Khan, Apoorva Bhattacharya, Shruti Banerjee, Swastika Paul, Abhishek Dutta, Anju Patel, Pankaj Kumar Singh, Shubho Chaudhuri and Amita Pal Dipanwita Dutta Chowdhury, Udit Basak, Apratim Dutta, Arijit Bhowmik, Devdutt A-2 Anupama Ghosh I Induction of apoptosis-like cell death and clearance of stress-induced intracellular Mazumdar, Aparajita Das, Sourio Chakraborty and Tanya Das protein aggregates: dual roles for Ustilago maydis metacaspase Mca1. E-1 Abhrajyoti Ghosh V Deciphering the code behind prokaryotic stress responses and ecophysiology A-1 Dibya Mukherjee, Sayandeep Gupta, Saran N, Rahul Datta, Anupama Ghosh Mousam Roy, Sayandeep Gupta, Chandrima Bhattacharyya, Shayantan Mukherji, A-3 Debabrata Basu I A multifaceted approach to unravel the signalling components of 'Black Spot' Abhrajyoti Ghosh A-2 disease resistance in oilseed mustard E-2 Srimonti Sarkar V The minimal ESCRT machinery of Giardia lamblia has altered inter-subunit Mrinmoy Mazumder, Amrita Mukherjee, Banani Mondal, Swagata Ghosh, Aishee De interactions within the ESCRT-II and ESCRT-III complexes A-3 and Debabrata Basu Nabanita Saha, Somnath Dutta and Srimonti Sarkar A-4 I Gaurab Gangyopadhyay Towards broadening the gene pool of few crop plants through molecular and E-3 Subrata Sau V Identification, purification and characterization of a cyclophilin from A-4 transgenic breeding Staphylococcus aureus Debabrata Dutta, Soumili Pal, Marufa Sultana, Vivek Arora and Gaurab Soham Seal, Debabrata Sinha, Subrata Sau Gangopadhyay E-4 Sujoy Kr. -

PE PGRS33 Contributes to Mycobacterium Tuberculosis Entry in Macrophages Through Interaction with TLR2
RESEARCH ARTICLE PE_PGRS33 Contributes to Mycobacterium tuberculosis Entry in Macrophages through Interaction with TLR2 Ivana Palucci1, Serena Camassa1, Alessandro Cascioferro3, Michela Sali1, Saber Anoosheh3, Antonella Zumbo1, Mariachiara Minerva1, Raffaella Iantomasi1, Flavio De Maio1, Gabriele Di Sante2, Francesco Ria2, Maurizio Sanguinetti1, Giorgio Palù3, Michael J. Brennan4, Riccardo Manganelli3, Giovanni Delogu1* 1 Institute of Microbiology, Università Cattolica del Sacro Cuore, L.go A. Gemelli, 8–00168, Rome, Italy, 2 Institute of General Pathology, Università Cattolica del Sacro Cuore, L.go A. Gemelli, 8–00168, Rome, Italy, 3 Department of Molecular Medicine, University of Padua, Via A. Gabelli, 63–35121, Padua, Italy, 4 Aeras, Rockville (MD), United States of America * [email protected] Abstract PE_PGRS represent a large family of proteins typical of pathogenic mycobacteria whose members are characterized by an N-terminal PE domain followed by a large Gly-Ala repeat- OPEN ACCESS rich C-terminal domain. Despite the abundance of PE_PGRS-coding genes in the Myco- Citation: Palucci I, Camassa S, Cascioferro A, Sali bacterium tuberculosis (Mtb) genome their role and function in the biology and pathogene- M, Anoosheh S, Zumbo A, et al. (2016) PE_PGRS33 sis still remains elusive. In this study, we generated and characterized an Mtb H37Rv Contributes to Mycobacterium tuberculosis Entry in mutant (MtbΔ33) in which the structural gene of PE_PGRS33, a prototypical member of the Macrophages through Interaction with TLR2. PLoS ONE 11(3): e0150800. doi:10.1371/journal. protein family, was inactivated. We showed that this mutant entered macrophages with an pone.0150800 efficiency up to ten times lower than parental or complemented strains, while its efficiency in Editor: Joyoti Basu, Bose Institute, INDIA infecting pneumocytes remained unaffected. -

IL-32: an Emerging Player in the Immune Response Network Against Tuberculosis? Manikuntala Kundu, Joyoti Basu*
Perspectives IL-32: An Emerging Player in the Immune Response Network against Tuberculosis? Manikuntala Kundu, Joyoti Basu* nterleukin-32 (IL-32), which was Stimulation of IL-32 by Strengths and Weaknesses previously called natural killer Mycobacteria of the Study cell transcript 4, has recently been I Netea et al. explored the role of IL- The crucial role of IFN-γ in recognized as a proinfl ammatory 32 in the context of M. tuberculosis antimycobacterial immunity has cytokine (see Glossary) [1]. The main infection. In their study, freshly been shown in patients with defects sources of IL-32 are natural killer obtained human peripheral blood in the IFN-γ receptor [7]. Netea and cells, T cells, epithelial cells, and mononuclear cells (PBMCs) were colleagues’ fi ndings raise important blood monocytes. Four transcripts of stimulated with different TLR agonists, questions regarding (a) how IL-32 IL-32 are known at present. IL-32 has and gene expression and synthesis of levels vary in patients with defects in emerged as an important player in IL-32 was determined. The authors IFN-γ receptors who are susceptible innate and adaptive immune responses, showed that M. tuberculosis could elicit to tuberculosis and (b) how these and information is emerging on IL-32 release from PBMCs as well as variations may impact the course of synergism between IL-32 and other from purifed monocyte populations. the infection. One of the defi ciencies well-characterized players in innate of their study stems from the fact immunity. that the role of IL-32 in controlling The innate immune response M. -

O509 2-Hour Oral Session Frontiers in Tuberculosis the Role of Micrornas
O509 2-hour Oral Session Frontiers in tuberculosis The role of microRNAs in tuning the immune response in Mycobacterium tuberculosis infection Joyoti Basu*1 1Bose Institute, Chemistry, Kolkata, India Background: The role of post-transcriptional regulatory mechanisms in calibrating the immune response of macrophages infected with Mycobacterium tuberculosis (Mtb), remains poorly understood. We have performed global miRNA and transcriptome profiling to build an miRNA – mRNA network in Mtb- infected macrophages, and to understand its impact on the immune response to infection. Material/methods: We have performed global qRT-PCR profiling of miRNAs and microarray analysis of mRNAs to profile the transcriptome of macrophages infected with Mtb. Bioinformatic analyses has been performed using the tool MAGIA, to build an miRNA-mRNA network. We have then focused on selected miRNAs to analyze their effects on the immune response by analyzing pathways associated with inflammation and autophagy. Results: During Mtb infection, the let-7 miRNAs appeared as highly connected nodes of the miRNA- mRNA network. We establish that A 20 (TNFAIP3), a negative regulator of NF- B signaling is a target of let- 7. Augmented release of TNF-alfa, IL-1beta, CCL2, CXCL1, IL-6 and NO occurs in Mtb- infected macrophages in the absence of A20. During infection, downregulation of let -7f leads to concomitant rise in A20, likely tuning down inflammatory mediators. Transfection with a let -7f mimic, leads to increased cytokine release after infection. In addition to let-7f, miR-17 family members are also downregulated during Mtb infection of macrophages. We show that forced expression of miR-17 enhances autophagy during Mtb infection. -

Year Book 2018 Year Book 2018
YEAR BOOK 2018 YEAR BOOK 2018 WEST BENGAL ACADEMY OF SCIENCE AND TECHNOLOGY CSIR-Indian Institute of Chemical Biology Jadavpur YEAR BOOK Kolkata 700 032 Registered under the West Bengal Act XXVI of 1961 (S/65001 of 1990-91) 2018 PAN – AAATW0707E Published by : Prof. Satyabrata Pal, Elected Member, ISI, FRSS Formerly, Dean, Post Graduate Studies, BCKV and Honorary Visiting Professor, ISI, Kolkata Editor, West Bengal Acadepmy of Science and Technology Assisted by : Dr. Arun Bandyopadhyay, Ph.D. Chief Scientist, CSIR-IICB, Kolkata-700 032 Secretary, West Bengal Academy of Science and Technology WAST Secretariat CSIR-Indian Institute of Chemical Biology 4, Raja S. C. Mullick Road WEST BENGAL Jadavpur, Kolkata 700 032 A C Telephone: (033) 2499-5796 A W A D e-mail: [email protected] E M Website: http://www.iicb.res.in/wast/index.html S T Y SCIENCE Printed by : WEST BENGAL ACADEMY OF SCIENCE AND TECHNOLOGY Creative Data Centre Registered Office : CSIR-Indian Institute of Chemical Biology 58/32, Prince Anwar Shah Road 4, Raja S. C. Mullick Road, Jadavpur Kolkata- 700 045 Kolkata 700 032 E-mail: [email protected] 1 2 YEAR BOOK 2018 YEAR BOOK 2018 AD-HOC Committee (1986-1989) Contents 1. Professor Sushil Kumar Mukherjee : Chairman 2. Professor Syama Pada Sen Introduction 5 3. Professor Asok Ghosh Memorandum of Association 6 4. Dr. Satyesh Chandra Pakrashi Rules and Regulations 9 Approved Amendments–I 25 5. Professor Subodh Kumar Roy Approved Amendments–II 29 6. Professor Asok Kumar Barua Past Office Bearers 34 7. Professor Nityananda Saha Council : 2016-2018 37 Sectional Committees : 2016-2018 39 8. -

Mycobacterial Signaling Through Toll-Like Receptors
MINI REVIEW ARTICLE published: 23 November 2012 CELLULAR AND INFECTION MICROBIOLOGY doi: 10.3389/fcimb.2012.00145 Mycobacterial signaling through toll-like receptors Joyoti Basu 1, Dong-Min Shin 2 and Eun-Kyeong Jo 2* 1 Department of Chemistry, Bose Institute, Kolkata, India 2 Department of Microbiology and Infection Signaling Network Research Center, Chungnam National University School of Medicine, Daejeon, South Korea Edited by: Studies over the past decade have helped to decipher molecular networks dependent Nelson Gekara, Umea University, on Toll-like receptor (TLR) signaling, in mycobacteria-infected macrophages. Stimulation Sweden of TLRs by mycobacteria and their antigenic components rapidly induces intracellular Reviewed by: signaling cascades involved in the activation of nuclear factor-κB and mitogen-activated Maximiliano G. Gutierrez, Medical Research Council- National Institute protein kinase pathways, which play important roles in orchestrating proinflammatory for Medical Research, UK responses and innate defense through generation of a variety of antimicrobial effector Javeed Ali Shah, University of molecules. Recent studies have provided evidence that mycobacterial TLR-signaling cross Washington, USA talks with other intracellular antimicrobial innate pathways, the autophagy process and *Correspondence: functional vitamin D receptor (VDR) signaling. In this article we describe recent advances Eun-Kyeong Jo, Department of Microbiology and Infection Signaling in the recognition, responses, and regulation of mycobacterial signaling through TLRs. Network Research Center, Chungnam National University Keywords: mycobacteria, vitamin D, autophagy, antimicrobial peptides, innate immunity School of Medicine, 301-747, Daejeon, South Korea. e-mail: [email protected] INTRODUCTION intracellular microbes, including Mtb (Deretic, 2012; Mintern Tuberculosis remains a serious health problem worldwide, caus- and Villadangos, 2012). -

Execution of Macrophage Apoptosis by Mycobacterium Avium Through
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 29, Issue of July 18, pp. 26517–26525, 2003 © 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. Execution of Macrophage Apoptosis by Mycobacterium avium through Apoptosis Signal-regulating Kinase 1/p38 Mitogen-activated Protein Kinase Signaling and Caspase 8 Activation* Received for publication, January 27, 2003, and in revised form, April 24, 2003 Published, JBC Papers in Press, April 30, 2003, DOI 10.1074/jbc.M300852200 Asima Bhattacharyya, Shresh Pathak, Chaitali Basak, Sujata Law, Manikuntala Kundu‡, and Joyoti Basu§ From the Department of Chemistry, Bose Institute, 93/1 Acharya Prafulla, Chandra Road, Kolkata 700 009, India Macrophage apoptosis is an important component of receptors. This results in subsequent internalization of the the innate immune defense machinery (against patho- mycobacterium within the macrophage. The macrophage rep- genic mycobacteria) responsible for limiting bacillary resents the first line in innate immune defense against myco- viability. However, little is known about the mechanism bacteria. In macrophages infected with pathogenic mycobacte- of how apoptosis is executed in mycobacteria-infected ria, apoptotic death is associated with a reduction in bacillary macrophages. Apoptosis signal-regulating kinase 1 viability (5–8). On the other hand, the success of virulent (ASK1) was activated in Mycobacterium avium-treated mycobacteria as pathogens is attributed largely to their ability Downloaded from macrophages and in turn activated p38 mitogen-acti- to survive and multiply within macrophages for extended pe- vated protein (MAP) kinase. M. avium-induced macro- riods of time. Mycobacteria live and proliferate within phago- phage cell death could be blocked in cells transfected somes, which unlike those containing other bacteria, are ar- with a catalytically inactive mutant of ASK1 or with rested in their maturation (9–11), lack lysosomal markers (12) dominant negative p38 MAP kinase arguing in favor of a and fail to be acidified (13). -

Awardees of National Bioscience Award for Career Development
AWARDEES OF NATIONAL BIOSCIENCE AWARD FOR CAREER DEVELOPMENT Awardees for the year 2016 1. Dr. Mukesh Jain, Associate Professor, School of Computational and Integrative Sciences, Jawaharlal Nehru University, New Delhi-110067 2. Dr. Samir K. Maji, Associate Professor, Indian Institute of Technology, Powai, Mumbai- 400076 3. Dr. Anindita Ukil, Assistant Professor, Calcutta University, Kolkata 4. Dr. Arnab Mukhopadhyay, Staff Scientist V, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi- 110067 5. Dr. Rohit Srivastava, Professor, Indian Institute of Technology, Bombay, Mumbai- 400076 6. Dr. Pinaki Talukdar, Associate Professor, Indian Institute of Science Education and Research, Dr. Homi Bhabha Road, Pashan, Pune- 7. Dr. Rajnish Kumar Chaturvedi, Senior Scientist, CSIR- Indian Institute of Toxicology Research, Lucknow-226001 8. Dr. Jackson James, Scientist E-II, Neuro Stem Cell Biology Lab, Neurobiology Division, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram, Kerala- 695014 Awardees for the year 2015 1. Dr. Sanjeev Das, Staff Scientist-V, National Institute of Immunology, New Delhi 2. Dr. Ganesh Nagaraju, Assistant Professor, Department of Biotechnology, Indian Institute of Science, Bangalore- 5600012. 3. Dr. Suvendra Nath Bhattacharya, Principal Scientist, CSIR- Indian Institute of Chemical Biology, Kolkata- 700032 4. Dr. Thulasiram H V, Principal Scientist, CSIR-National Chemical Laboratory, Pune- 411008. 5. Dr. Pawan Gupta, Principal Scientist, Institute of microbial Technology, Chandigarh- 160036. 6. Dr. Souvik Maiti, Principal Scientist, CSIR-Institute of Genomics and Integrative Biology, Delhi- 110025. 7. Dr. Pravindra Kumar, Associate Professor, Department of Biotechnology, IIT, Roorkee- 247667. 8. Dr. Anurag Agrawal, Principal Scientist, CSIR-Institute of Genomics and Integrative Biology, Delhi- 110025 9. Dr. Gridhar Kumar Pandey, Professor, Department of Plant Molecular Biology, University of Delhi South Campus, New Delhi- 110067 10. -

National Bioscience Awards for Career Development
AWARDEES OF NATIONAL BIOSCIENCE AWARDS FOR CAREER DEVELOPMENT Awardees for the year 2012 1. Dr. Kaustuv Sanyal, Associate Professor, Molecular Mycology Laboratory, Molecular Biology & Genetics Unit, Jawaharlal Nehru Centre for Advance Scientific Research, Jakkur P.O. Bangalore 560064 2. Dr Naval Kishore Vikram, Associate Professor, Department of Medicine, All India Institute of Medical Sciences (AIIMS), Ansari Nagar, New Delhi- 110029 3. Dr. Aditya Bhushan Pant, Senior Scientist & In-charge, In Vitro Toxicology Laboratory, Indian Institute of Toxicology Research, PO Box: 80, MG Marg, Lucknow 226001 (UP) India 4. Dr. Subrata Adak, Senior Scientist, Indian Institute of Chemical Biology; 4, Raja S.C. Mullick Road, Kolkata-700032 5. Dr. Durai Sundar, Assistant Professor, Dept of Biochemical Engineering & Biotechnology, Indian Institute of Technology (IIT) Delhi, Hauz Khas, New Delhi – 110016 6. Dr S Venkata Mohan, Senior Scientist, Bioengineering and Environmental Centre (BEEC) CSIR-Indian Institute of Chemical Technology, Hyderabad-500 607 7. Dr. Munia Ganguli, Scientist E-I, CSIR-Institute of Genomics & Integrative Biology, Mall Road,New Delhi 110 007 8. Dr. Asad U Khan, Associate Professor & Coordinator/Head of Biotechnology Department, A.M.U, Interdisciplinary Biotechnology Unit, A.M.U., Aligarh 202002 9. Dr. Sathees C. Raghavan, Assistant Professor, Department of Biochemistry, Indian Institute of Science, Bangalore 560 012 10. Dr. Vidita A. Vaidya, Associate Professor, Department of Biological Sciences, Tata Institute of Fundamental Research, 1, Homi Bhabha Road, Colaba, Mumbai - 400005 Awardees for the year 2011 1. Dr. M. M. Parida, Scientist-F, Joint Director, Division of Virology Defence R & D Establishment, DRDE, DRDO, Ministry of Defence, Jhansi Road, Gwalior- 474002 2. -

Differential Targeting of C-Maf, Bach-1, and Elmo-1 by Microrna
ORIGINAL RESEARCH published: 19 March 2019 doi: 10.3389/fimmu.2019.00421 Differential Targeting of c-Maf, Bach-1, and Elmo-1 by microRNA-143 and microRNA-365 Promotes the Intracellular Growth of Mycobacterium tuberculosis in Alternatively IL-4/IL-13 Activated Macrophages Edited by: Jagadeesh Bayry, 1,2,3 1,2 1,2 1,2 Institut National de la Santé et de la Ousman Tamgue , Lorna Gcanga , Mumin Ozturk , Lauren Whitehead , 1,2 1,2 4 5 Recherche Médicale (INSERM), Shandre Pillay , Raygaana Jacobs , Sugata Roy , Sebastian Schmeier , France Malika Davids 6, Yulia A. Medvedeva 7,8,9, Keertan Dheda 6,10, Harukazu Suzuki 4, 1,2,11 1,2,11 Reviewed by: Frank Brombacher * and Reto Guler * Sahana Holla, 1 International Centre for Genetic Engineering and Biotechnology, Cape Town Component, Cape Town, South Africa, National Cancer Institute (NCI), 2 Division of Immunology and South African Medical Research Council Immunology of Infectious Diseases, Department of United States Pathology, Faculty of Health Sciences, Institute of Infectious Diseases and Molecular Medicine, University of Cape Town, Joyoti Basu, Cape Town, South Africa, 3 Department of Biochemistry, Faculty of Sciences, University of Douala, Douala, Cameroon, Bose Institute, India 4 RIKEN Center for Integrative Medical Sciences, Yokohama, Japan, 5 Institute of Natural and Mathematical Sciences, Massey *Correspondence: University, Auckland, New Zealand, 6 Centre for Lung Infection and Immunity, Department of Medicine and UCT Lung Frank Brombacher Institute, Division of Pulmonology, University -

Macrophage Control of Phagocytosed Mycobacteria Is Increased by Factors Secreted by Alveolar Epithelial Cells Through Nitric Oxide Independent Mechanisms
Macrophage Control of Phagocytosed Mycobacteria Is Increased by Factors Secreted by Alveolar Epithelial Cells through Nitric Oxide Independent Mechanisms Dagbjort H. Petursdottir*., Olga D. Chuquimia., Raphaela Freidl, Carmen Ferna´ndez Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, Stockholm, Sweden Abstract Tissue-resident macrophages are heterogeneous with tissue-specific and niche-specific functions. Thus, simplified models of macrophage activation do not explain the extent of heterogeneity seen in vivo. We focus here on the respiratory tract and ask whether factors secreted by alveolar epithelial cells (AEC) can influence the functionality of resident pulmonary macrophages (PuM). We have previously reported that factors secreted by AEC increase control of intracellular growth of BCG in macrophages. In the current study, we also aimed to investigate possible mechanisms by which AEC-derived factors increase intracellular control of BCG in both primary murine interstitial macrophages, and bone marrow-derived macrophages and characterize further the effect of these factors on macrophage differentiation. We show that; a) in contrast to other macrophage types, IFN-c did not increase intracellular growth control of Mycobacterium bovis, Bacillus Calmette-Gue´rin (BCG) by interstitial pulmonary macrophages although the same macrophages could be activated by factors secreted by AEC; b) the lack of response of pulmonary macrophages to IFN-c was apparently regulated by suppressor of cytokine signaling (SOCS)1; c) AEC-derived factors did not induce pro-inflammatory pathways induced by IFN- c e.g. expression of inducible nitric oxide synthase (iNOS), secretion of nitric oxide (NO), or IL-12, d) in contrast to IFN-c, intracellular bacterial destruction induced by AEC-derived factors was not dependent on iNOS transcription and NO production.