2'-5'-Oligoadenylate Synthetase 1 (OAS1) and Health Disparities in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Genetic Variant Protective Against Severe COVID-19 Is Inherited from Neandertals
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.05.327197; this version posted October 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A genetic variant protective against severe COVID-19 is inherited from Neandertals Authors Hugo Zeberg1,2* and Svante Pääbo1,3* Affiliations 1 Max Planck Institute for Evolutionary Anthropology, Deutscher Platz 6, D-04103 Leipzig, Germany. 2 Department of Neuroscience, Karolinska Institutet, SE-17177 Stockholm, Sweden. 3 Okinawa Institute of Science and Technology, Onna-son, Okinawa 904-0495, Japan. *Corresponding authors: [email protected], [email protected] Abstract It was recently shown that the major genetic risk factor associated with becoming severely ill with COVID-19 when infected by SARS-CoV-2 is inherited from Neandertals. Thanks to new genetic association studies additional risk factors are now being discovered. Using data from a recent genome- wide associations from the Genetics of Mortality in Critical Care (GenOMICC) consortium, we show that a haplotype at a region associated with requiring intensive care is inherited from Neandertals. It encodes proteins that activate enzymes that are important during infections with RNA viruses. As compared to the previously described Neandertal risk haplotype, this Neandertal haplotype is protective against severe COVID-19, is of more moderate effect, and is found at substantial frequencies in all regions of the world outside Africa. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.05.327197; this version posted October 9, 2020. -

1A Multiple Sclerosis Treatment
The Pharmacogenomics Journal (2012) 12, 134–146 & 2012 Macmillan Publishers Limited. All rights reserved 1470-269X/12 www.nature.com/tpj ORIGINAL ARTICLE Network analysis of transcriptional regulation in response to intramuscular interferon-b-1a multiple sclerosis treatment M Hecker1,2, RH Goertsches2,3, Interferon-b (IFN-b) is one of the major drugs for multiple sclerosis (MS) 3 2 treatment. The purpose of this study was to characterize the transcriptional C Fatum , D Koczan , effects induced by intramuscular IFN-b-1a therapy in patients with relapsing– 2 1 H-J Thiesen , R Guthke remitting form of MS. By using Affymetrix DNA microarrays, we obtained and UK Zettl3 genome-wide expression profiles of peripheral blood mononuclear cells of 24 MS patients within the first 4 weeks of IFN-b administration. We identified 1Leibniz Institute for Natural Product Research 121 genes that were significantly up- or downregulated compared with and Infection Biology—Hans-Knoell-Institute, baseline, with stronger changed expression at 1 week after start of therapy. Jena, Germany; 2University of Rostock, Institute of Immunology, Rostock, Germany and Eleven transcription factor-binding sites (TFBS) are overrepresented in the 3University of Rostock, Department of Neurology, regulatory regions of these genes, including those of IFN regulatory factors Rostock, Germany and NF-kB. We then applied TFBS-integrating least angle regression, a novel integrative algorithm for deriving gene regulatory networks from gene Correspondence: M Hecker, Leibniz Institute for Natural Product expression data and TFBS information, to reconstruct the underlying network Research and Infection Biology—Hans-Knoell- of molecular interactions. An NF-kB-centered sub-network of genes was Institute, Beutenbergstr. -

Structural Basis for Cytosolic Double-Stranded RNA Surveillance by Human Oligoadenylate Synthetase 1
Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1 Jesse Donovan, Matthew Dufner, and Alexei Korennykh1 Department of Molecular Biology, Princeton University, Princeton, NJ 08540 Edited by Jennifer A. Doudna, University of California, Berkeley, CA, and approved December 19, 2012 (received for review October 23, 2012) The human sensor of double-stranded RNA (dsRNA) oligoadenylate Results and Discussion synthetase 1 (hOAS1) polymerizes ATP into 2′,5′-linked iso-RNA (2- Overview of the hOAS1•dsRNA•dATP Ternary Complex. To un- 5A) involved in innate immunity, cell cycle, and differentiation. We derstand how OAS1 recognizes dsRNA, we conducted cocrys- report the crystal structure of hOAS1 in complex with dsRNA and tallization screening with hOAS1 and dsRNA sequences derived 2′-deoxy ATP at 2.7 Å resolution, which reveals the mechanism of from RNA constructs known to activate the sensor (2). Single cytoplasmic dsRNA recognition and activation of oligoadenylate cocrystals were obtained only with dsRNA having 18 bp and a ser- synthetases. Human OAS1 recognizes dsRNA using a previously endipitously constructed sequence GGCUUUUGACCUUUAU- uncharacterized protein/RNA interface that forms via a conforma- GC. The structure of the ternary complex was determined at 2.7 Å tional change induced by binding of dsRNA. The protein/RNA in- resolution (Table S1). In the cocrystal structure, hOAS1 is bound to fi terface involves two minor grooves and has no sequence-speci c one face of the RNA double-helix (Fig. 1A and Figs. S1 and S2)and contacts, with the exception of a single hydrogen bond between the dsRNA termini are unobstructed by the protein (Table S2). -

The Evolution of the Viral RNA Sensor OAS1 in Old World Monkeys and Cetartiodactyls
City University of New York (CUNY) CUNY Academic Works All Dissertations, Theses, and Capstone Projects Dissertations, Theses, and Capstone Projects 2-2016 The Evolution of the Viral RNA Sensor OAS1 in Old World Monkeys and Cetartiodactyls Ian Fish Graduate Center, City University of New York How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/gc_etds/759 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] The Evolution of the Viral RNA Sensor OAS1 in Old World Monkeys and Cetartiodactyls by Ian Fish The City University of New York 2016 i Copyright 2016 by Fish, Ian All rights reserved ii This manuscript has been read and accepted for the Graduate Faculty in Biology in satisfaction of the dissertation requirement for the degree of Doctor of Philosophy. ______________ ______________________________ Date Chair of Examining Committee Dr. Stéphane Boissinot ______________ ______________________________ Date Executive Officer Dr. Laurel Eckhardt Supervising Committee Members: ____________________________ Dr. Cathy Savage-Dunn, Queens College ____________________________ Dr. Susan Rotenberg, Queens College ____________________________ Dr. Shaneen Singh, Brooklyn College ____________________________ Dr. Margaret MacDonald, The Rockefeller University iii Abstract The Evolution of the Viral RNA Sensor OAS1 in Old World Monkeys and Cetartiodactyls author: Ian Fish advisor: Dr. Stéphane Boissinot Animals produce an array of sensors patrolling the intracellular environment poised to detect and respond to viral infection. The oligoadenylate synthetase family of enzymes comprises a crucial part of this innate immune response, directly signaling endonuclease activity responsible for inhibiting viral replication. -

A Neanderthal OAS1 Isoform Protects Against COVID-19 Susceptibility
medRxiv preprint doi: https://doi.org/10.1101/2020.10.13.20212092; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . 1 A Neanderthal OAS1 isoform Protects Against COVID-19 Susceptibility and 2 Severity: Results from Mendelian Randomization and Case-Control Studies 3 4 5 Sirui Zhou1,2,* Guillaume Butler-Laporte1,2,* Tomoko Nakanishi1,3,4,5,* David Morrison1, Jonathan Afilalo1, 6 Marc Afilalo1, Laetitia Laurent1, Maik Pietzner6, Nicola Kerrison6, Kaiqiong Zhao1,2, Elsa Brunet- 7 Ratnasingham7, Danielle Henry1, Nofar Kimchi1, Zaman Afrasiabi1, Nardin Rezk1, Meriem Bouab1, Louis 8 Petitjean,1 Charlotte Guzman1, Xiaoqing Xue1, Chris Tselios,1 Branka Vulesevic1, Olumide Adeleye1, Tala 9 Abdullah1, Noor Almamlouk1, Yiheng Chen1, Michaël Chassé7, Madeleine Durand7, Michael Pollak1, Clare 10 Paterson8, Hugo Zeberg9, Johan Normark10, Robert Frithiof11, Miklós Lipcsey12,13, Michael Hultström11,13, 11 Celia M T Greenwood1,2, Claudia Langenberg6,14, Elin Thysell15, Vincent Mooser3, Vincenzo Forgetta1, 12 Daniel E. Kaufmann7,16, J Brent Richards1,2,3,17 13 14 Affiliations: 15 1) Lady Davis Institute, Jewish General Hospital, McGill University, Montréal, Québec, 16 Canada 17 2) Department of Epidemiology, Biostatistics and Occupational Health, McGill 18 University, Montréal, Québec, Canada 19 3) Department of Human Genetics, -

Supplementary Table 1: Gene List of 44 Upregulated Enzymes in Transformed Mesenchymal Stem Cell Cancer Model
Supplementary Table 1: Gene list of 44 upregulated enzymes in transformed mesenchymal stem cell cancer model. Gene expression values for parental MSC (MSC 0) and transformed MSC (MSC5) are an average of three replicate log-2 transformed expression values from affymetrix U133 plus 2 genechip experiments with the log-fold change (LFC) indicating the difference (MSC5-MSC0). Supplementary Table 1 HGNC Symbol Alias Enzyme ID U133 plus2 probe set MSC0 MSC5 LFC Ttest pval # Gene rifs # Pubmed cites from Genecards (Sep 2007) Pathway / Function PharmGKB Drugs? Drug pathways? Therapeutic Target Database Thomson Pharma RNASEH2A AGS4; JUNB; RNHL; RNHIA; RNASEHI 3.1.26.- 203022_at 8.56 9.87 1.31 1.56E-05 0 11 RNA degradation none none none PPAP2C LPP2; PAP-2c; PAP2-g 3.1.3.4 209529_at 6.48 8.63 2.16 5.74E-03 2 13 Glycerolipid synthesis none none none ADARB1 ADAR2, ADAR2a, ADAR2a-L1, ADAR2a-L2, ADAR2a-L3, ADAR2b, ADAR2c 3.5.-.- 234799_at 6.36 8.15 1.79 2.03E-04 10 58 RNA pre-mRNA editing none none none ADARB1 3.5.-.- 203865_s_at 6.99 8.42 1.43 6.94E-03 10 58 RNA pre-mRNA editing none none none UAP1 AgX; AGX1; SPAG2 2.7.7.23 209340_at 11.17 12.45 1.28 2.46E-07 0 37 polysaccharide synthesis none none RNMT MET; RG7MT1; hCMT1c; KIAA0398; DKFZp686H1252 2.1.1.56 202684_s_at 5.70 6.78 1.08 8.15E-03 1 24 RNA (mRNA) capping none none GPD2 GDH2, mGPDH 1.1.1.8 211613_s_at 5.71 6.73 1.02 7.02E-03 2 37 glycolysis none none GCDH ACAD5, GCD 1.3.99.7 237304_at 5.38 6.39 1.01 2.44E-02 4 63 lys, hydroxy-lys, and trp metabolism none none ESPL1 3.4.22.49 38158_at 8.03 -

Cancer Upregulated Gene 2, a Novel Oncogene, Confers Resistance to Oncolytic Vesicular Stomatitis Virus Through STAT1-OASL2 Signaling
Cancer Gene Therapy (2013) 20, 125–132 & 2013 Nature America, Inc. All rights reserved 0929-1903/13 www.nature.com/cgt ORIGINAL ARTICLE Cancer upregulated gene 2, a novel oncogene, confers resistance to oncolytic vesicular stomatitis virus through STAT1-OASL2 signaling W Malilas1, SS Koh2, R Srisuttee1, W Boonying1, I-R Cho1, C-S Jeong3, RN Johnston4 and Y-H Chung1 We have recently found a novel oncogene, named cancer upregulated gene 2 (CUG2), which activates Ras and mitogen-activated protein kinases (MAPKs), including ERK, JNK and p38 MAPK. Because activation of these signaling pathways has previously been shown to enhance cancer cell susceptibility to oncolysis by certain viruses, we examined whether vesicular stomatitis virus (VSV) could function as a potential therapeutic agent by efficiently inducing cytolysis in cells transformed by CUG2. Unexpectedly, NIH3T3 cells stably expressing CUG2 (NIH-CUG2) were resistant to VSV because of the activation of signal transducers and activators of transcription 1 (STAT1). The result was supported by evidence showing that suppression of STAT1 with short interference RNA (siRNA) renders cells susceptible to VSV. Furthermore, 20–50 oligoadenylate synthetase-like (OASL) 2 was the most affected by STAT1 expression level among anti-viral proteins and furthermore suppression of OASL2 mRNA level caused NIH-CUG2 cells to succumb to VSV as seen in NIH-CUG2 cells treated with STAT1 siRNA. In addition, Colon26L5 carcinoma cells stably expressing CUG2 (Colon26L5-CUG2) exhibited resistance to VSV, whereas Colon26L5 stably expressing a control vector yielded to VSV infection. Moreover, Colon26L5-CUG2 cells stably suppressing STAT1 succumbed to VSV infection, resulting in apoptosis. -

14498 OAS1 (D1W3A) Rabbit Mab
Revision 1 C 0 2 - t OAS1 (D1W3A) Rabbit mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] 8 Support: 877-678-TECH (8324) 9 4 Web: [email protected] 4 www.cellsignal.com 1 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB, IP H Endogenous 40, 44 Rabbit IgG P00973 4938 Product Usage Information Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:50 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity OAS1 (D1W3A) Rabbit mAb recognizes endogenous levels of total OAS1 protein. This antibody cross-reacts with an unidentified protein of 100 kDa in some cell lines. Species Reactivity: Human Source / Purification Monoclonal antibody is produced by immunizing animals with a synthetic peptide corresponding to residues surrounding Asp90 of human OAS1 protien. Background 2’-5’-oligoadenylate synthetase 1 (OAS1) is an antiviral protein induced by type 1 interferon that plays a key role in the cellular innate immune response (1). The OAS family of proteins includes OAS1, OAS2, OAS3, and OASL in humans (2). The OAS1 enzyme produces the second messenger 2’-5’-linked oligoadenylate in response to cytosolic dsRNA. These 2’-5’-linked oligoadenylates bind to the ribonuclease RNase L, which then degrades viral and cellular RNA (3). Research studies indicate that the OAS1 system inhibits protein synthesis and induces apoptosis in virally infected cells, which limits viral infection (4). -

A Prenylated Dsrna Sensor Protects Against Severe COVID-19 and Is Absent in Horseshoe Bats
medRxiv preprint doi: https://doi.org/10.1101/2021.05.05.21256681; this version posted May 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . @WilsonlabCVR Wickenhagen et al., A Prenylated dsRNA Sensor Protects Against Severe COVID-19 and is ABsent in Horseshoe Bats A Prenylated dsRNA Sensor Protects on global health, culture and prosperity. SARS-CoV-2 is Against Severe COVID-19 and is Absent in particularly sensitive to inhibition by type I interferons (IFNs) and there is great interest in understanding how individual Horseshoe Bats IFN-stimulated genes (ISGs) inhibit SARS-CoV-2, as type I IFNs play a major role in governing COVID-19 disease Arthur Wickenhagen1, Elena Sugrue1*, Spyros Lytras1*, 1* 1 1 outcome. Specifically, inborn errors (Zhang et al., 2020) Srikeerthana Kuchi , Matthew L Turnbull , Colin Loney , and single nucleotide polymorphisms (Pairo-Castineira et 1 1 1 Vanessa Herder , Jay Allan , Innes Jarmson , Natalia al., 2020) within the IFN system are linked with more severe 1 1 1 Cameron-Ruiz , Margus Varjak , Rute M Pinto , Douglas G COVID-19. Moreover, neutralizing anti-IFN autoantibodies 1 1 1 Stewart , Simon Swingler , Marko Noerenberg , Edward J likely prevent host IFN responses from controlling SARS- 2 2 1 D Greenwood , Thomas W M Crozier , Quan Gu , Sara CoV-2 replication, again resulting in severe COVID-19 3 3 4 Clohisey , Bo Wang , Fabio Trindade Maranhão Costa , disease (Bastard et al., 2020). -

Real-Time 2-5A Kinetics Suggest That Interferons Β and Λ Evade Global Arrest of Translation by Rnase L
Real-time 2-5A kinetics suggest that interferons β and λ evade global arrest of translation by RNase L Alisha Chitrakara,1, Sneha Ratha,1, Jesse Donovana, Kaitlin Demaresta, Yize Lib, Raghavendra Rao Sridharc,d, Susan R. Weissb, Sergei V. Kotenkoc,d, Ned S. Wingreena, and Alexei Korennykha,2 aDepartment of Molecular Biology, Princeton University, Princeton, NJ 08544; bDepartment of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; cDepartment of Microbiology, Biochemistry, and Molecular Genetics, Rutgers New Jersey Medical School, Newark, NJ 07103; and dCenter for Immunity and Inflammation, Rutgers New Jersey Medical School, Newark, NJ 07103 Edited by Peter Walter, University of California, San Francisco, CA, and approved December 14, 2018 (received for review October 24, 2018) Cells of all mammals recognize double-stranded RNA (dsRNA) as a of a dimeric endoribonuclease active site (21–23). This dimer foreign material. In response, they release interferons (IFNs) and further assembles into high-order oligomers (24) that cleave viral activate a ubiquitously expressed pseudokinase/endoribonuclease RNAs (16, 18) and all components of the translation apparatus, RNase L. RNase L executes regulated RNA decay and halts global including mRNAs (25), tRNAs (26), and 28S/18S rRNAs (27, translation. Here, we developed a biosensor for 2′,5′-oligoadeny- 28). The resulting action of RNase L inhibits global translation, late (2-5A), the natural activator of RNase L. Using this biosensor, which puts all proteins, including IFNs, at risk for arrest during a we found that 2-5A was acutely synthesized by cells in response to cellular response to dsRNA. dsRNA sensing, which immediately triggered cellular RNA cleav- The impact of translational shutdown by RNase L on IFN age by RNase L and arrested host protein synthesis. -
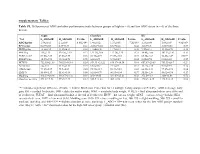
Supplementary Tables: Table S1
Supplementary Tables: Table S1. Differences of ADG and other performance traits between groups of high (n = 6) and low ADG steers (n = 6) of the three breeds. Angus Charolais KC Trait L_ADG±SE H_ADG±SE P-value L_ADG±SE H_ADG±SE P-value L_ADG±SE H_ADG±SE P-value ADG/kg/day 1.54±0.02 2.1±0.09 8.06E-04* 1.48±0.02 1.93±0.05 7.2E-05* 1.23±0.04 1.88±0.09 4.8E-05* RFI/kg/day 0.63±0.35 0.37±0.39 0.63 -0.032±0.40 0.33±0.28 0.48 -0.65±0.3 -0.06±0.56 0.37 DMI/kg/day 12.2±0.34 13.45±0.34 0.03 11.06±0.48 11.7±0.34 0.30 9.57±0.31 11.12±0.78 0.10 MWT/kg 115±2.33 120.26±2.19 0.13 122.31±2.06 117.76±2.25 0.22 98.45±1.66 103.53±3.02 0.17 FUREA/cm2 83.08±2.89 82.88±2.95 0.96 94.78±4.91 91.59±3.83 0.73 65.34±1.62 74.45±1.49 0.002* FUFAT/mm 10.57±0.93 10.18±0.98 0.78 6.38±0.71 5.73±0.37 0.44 8.28±0.94 8.83±0.32 0.59 HCW/lb 752.95±21.68 786.53±16.14 0.24 859.33 ±21.22 836.17±16.34 0.41 657.67±20.83 703.33±26.7 0.21 AFAT/mm 12±1.13 11.33±1.31 0.71 6.67±0.67 7.17±0.4 0.53 11.17±1.78 9.83±0.95 0.52 CREA/cm2 73.17±2.87 75.5±4.09 0.65 95.33±3.17 90.33±5.18 0.43 66.83±3.33 77.17±2.98 0.04 LMY/% 55.08±1.17 55.61±1.66 0.80 62.01±0.79 60.93±0.84 0.37 55.55±1.36 58.02±0.94 0.17 Marbling 448.33±24.96 416.67±24.32 0.39 365±19.05 381.67±25.23 0.58 372.5±9.64 380±18.08 0.72 Slaughter age/day 491.67±7.52 493.5±4.43 0.84 525.5 ±1.28 504±8.98 0.06 470.83±8.35 448.5±6.14 0.06 “*” indicates significant difference (P-value < 0.0042, Bonferroni Correction for 12 multiple testing analyses at P<0.05). -

IL36RN Mutations
The University of Manchester Research IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis DOI: 10.1016/j.jid.2019.08.444 Document Version Accepted author manuscript Link to publication record in Manchester Research Explorer Citation for published version (APA): Catapano, M., Vergnano, M., Romano, M., Mahil, S. K., Choon, S., Burden, A. D., Young, H. S., Carr, I. M., Lachmann, H. J., Lombardi, G., Smith, C. H., Ciccarelli, F. D., Barker, J. N., & Capon, F. (2019). IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis. Journal of Investigative Dermatology. https://doi.org/10.1016/j.jid.2019.08.444 Published in: Journal of Investigative Dermatology Citing this paper Please note that where the full-text provided on Manchester Research Explorer is the Author Accepted Manuscript or Proof version this may differ from the final Published version. If citing, it is advised that you check and use the publisher's definitive version. General rights Copyright and moral rights for the publications made accessible in the Research Explorer are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Takedown policy If you believe that this document breaches copyright please refer to the University of Manchester’s Takedown Procedures [http://man.ac.uk/04Y6Bo] or contact [email protected] providing relevant details, so we can investigate your claim. Download date:03.