Recent Advances in the Synthesis of 2-Pyrones
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proquest Dissertations
The synthesis of highly substituted indoles via isonitriles Item Type text; Dissertation-Reproduction (electronic) Authors Kennedy, Abigail Rose Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 26/09/2021 04:52:53 Link to Item http://hdl.handle.net/10150/290368 INFORMATION TO USERS This manuscript has l)een reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be firom any type of computer printer. The quality of this raproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author dkJ not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sectkms with small overiaps. Photographs included in the original manuscript have been reproduced xerographicaliy in this copy. Higher quality 6' x 9* black and white photographk: prints are available for any photographs or illustrations appearing in this copy for an additk)nai charge. -

Efficient Stille Cross-Coupling Reaction Catalyzed by the Pd(Oac
Efficient Stille Cross-Coupling Reaction found that Dabco was a highly efficient ligand for the - Catalyzed by the Pd(OAc)2/Dabco Catalytic palladium-catalyzed Suzuki Miyaura cross-coupling re- System action resulting in high TONs (up to 950 000 TONs for the reaction of PhI and p-chlorophenylboronic acid) (eq 7,8 Jin-Heng Li,* Yun Liang, De-Ping Wang, Wen-Jie Liu, 1). Thus, we expected to extend the use of this type of Ye-Xiang Xie, and Du-Lin Yin ligand to other palladium-catalyzed transformations. Here, we report a stable, inexpensive, and efficient Key Laboratory of Chemical Biology & Traditional Pd(OAc)2/Dabco catalytic system for the Stille reactions Chinese Medicine Research, College of Chemistry and of aryl halides with organotin compounds. Chemical Engineering, Hunan Normal University, Changsha 410081, China [email protected] Received October 31, 2004 To evaluate the efficiency of Pd(OAc)2/Dabco in the Stille cross-coupling reaction, coupling of 4-bromoanisole (1a) with phenyltributyltin (2a) was first tested, and the An efficient Pd(OAc)2/Dabco-catalyzed Stille cross-coupling reaction procedure has been developed. In the presence of (2) For recent representative papers on phosphine-palladium cata- Pd(OAc)2 and Dabco (triethylenediamine), various aryl lysts, see: (a) Litter, A. F.; Fu, G. C. Angew. Chem., Int. Ed. 1999, 38, halides including aryl iodides, aryl bromides, and activated 2411. (b) Grasa, G. A.; Nolan, S. P. Org. Lett. 2001, 3, 119. (c) Litter, aryl chlorides were coupled efficiently with organotin com- A. F.; Schwarz, L.; Fu, G. C. J. -

Stille and Suzuki-Miyaura Reactions
molecules Article Recoverable Palladium-Catalyzed Carbon-Carbon Bond Forming Reactions under Thermomorphic Mode: Stille and Suzuki-Miyaura Reactions Eskedar Tessema 1,†, Vijayanath Elakkat 1,† , Chiao-Fan Chiu 2,3,*, Zong-Lin Tsai 1, Ka Long Chan 1 , Chia-Rui Shen 4,5, Han-Chang Su 6 and Norman Lu 1,7,* 1 Institute of Organic and Polymeric Materials, National Taipei University of Technology, Taipei 106, Taiwan; [email protected] (E.T.); [email protected] (V.E.); [email protected] (Z.-L.T.); [email protected] (K.L.C.) 2 Department of Pediatrics, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 3 Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan 4 Department of Medical Biotechnology and Laboratory Sciences, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan; [email protected] 5 Department of Ophthalmology, Linkou Medical Center, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan 6 Creditable Service Technology Consultants, New Taipei City 235, Taiwan; [email protected] 7 Development Center for Smart Textile, National Taipei University of Technology, Taipei 106, Taiwan * Correspondence: [email protected] (C.-F.C.); [email protected] (N.L.). † These authors contributed equally to this work. Citation: Tessema, E.; Elakkat, V.; 0 0 Chiu, C.-F.; Tsai, Z.-L.; Chan, K.L.; Abstract: The reaction of [PdCl2(CH3CN)2] and bis-4,4 -(RfCH2OCH2)-2,2 -bpy (1a–d), where Rf = n- Shen, C.-R.; Su, H.-C.; Lu, N. C11F23 (a), n-C10F21 (b), n-C9F19 (c) and n-C8F17 (d), respectively, in the presence of dichloromethane 0 0 Recoverable Palladium-Catalyzed (CH2Cl2) resulted in the synthesis of Pd complex, [PdCl2[4,4 -bis-(RfCH2OCH2)-2,2 -bpy] (2a–d). -

Misassigned Natural Products and the Role of Chemical Synthesis in Modern Structure Elucidation K
Reviews K. C. Nicolaou and S. A. Snyder Natural Products Synthesis Chasing Molecules That Were Never There: Misassigned Natural Products and the Role of Chemical Synthesis in Modern Structure Elucidation K. C. Nicolaou* and Scott A. Snyder Keywords: azaspiracid-1 · diazonamide A · natural products · revised structures · total synthesis Angewandte Chemie 1012 2005 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim DOI: 10.1002/anie.200460864 Angew. Chem. Int. Ed. 2005, 44, 1012 – 1044 Angewandte Natural Products Synthesis Chemie Over the course of the past half century, the structural elucidation of From the Contents unknown natural products has undergone a tremendous revolution. Before World War II, a chemist would have relied almost exclusively 1. Introduction 1013 on the art of chemical synthesis, primarily in the form of degradation 2. The State of Modern Structure and derivatization reactions, to develop and test structural hypotheses Elucidation 1015 in a process that often took years to complete when grams of material were available. Today, a battery of advanced spectroscopic methods, 3. The Ramifications of Structural such as multidimensional NMR spectroscopy and high-resolution Misassignments 1026 mass spectrometry, not to mention X-ray crystallography, exist for the 4. Misassignment Case Studies 1029 expeditious assignment of structures to highly complex molecules isolated from nature in milligram or sub-milligram quantities. In fact, 5. Summary and Outlook 1037 it could be argued that the characterization of natural products has become a routine task, one which no longer even requires a reaction flask! This Review makes the case that imaginative detective work and chemical synthesis still have important roles to play in the process of solving natures most intriguing molecular puzzles. -

Regioselectivity Reaction Analytics
Regioselectivity: An Application of Expert Systems and Ontologies to Chemical (Named) Reaction Analysis Roger Sayle, John Mayfield and Noel O’Boyle NextMove Software, Cambridge, UK CINF Reaction Analytics. 256th ACS National Meeting, Boston, MA. Thursday 23rd August 2018 analysis vs. prediction • In “Applied Chemoinformatics” (2018), J. Goodman defines three main problems. – Reaction Planning: R → ? → P [Database] – Reaction Prediction: R1 + R2 → ? [Simulation] – Synthesis Planning: R? + R? + R? → P [Design] • A corollary is that there’s a distinction between reactions that have already been observed and those experiments yet to be performed. CINF Reaction Analytics. 256th ACS National Meeting, Boston, MA. Thursday 23rd August 2018 analysis vs. prediction CINF Reaction Analytics. 256th ACS National Meeting, Boston, MA. Thursday 23rd August 2018 intuition and counter-intuition With apologies to Mark Twain: “Never let the facts get in the way of a good synthesis plan”. There are reactions that chemists expect will happen but don’t & those they don’t expect but do. But cheminformaticians are alchemists that can turn lead into gold, as easily as “[Pb]>>[Au]”. CINF Reaction Analytics. 256th ACS National Meeting, Boston, MA. Thursday 23rd August 2018 Experimental validation • Synthesis of a novel aromatic heterocycle previously unreported in the literature. • William Pitt et al., “Heteroaromatic Rings of the Future”, Journal of Medicinal Chemistry, 52(9):2952- 2963, 2009. CINF Reaction Analytics. 256th ACS National Meeting, Boston, MA. Thursday 23rd August 2018 expectations: setting the scope Goodman Challenges Carey et al. Challenges Maitotoxin Difficult to access substituted aromatic starting materials. Eribulin Org. Biomol. Chem. 2005, 4,2337-2347. CINF Reaction Analytics. -

Cross-Coupling Reactions: A
Joesphine Eshon & Prithvi Vangal 1 11th November 2014 . Introduction . Cross-coupling reactions: A. Negishi* Reaction B. Heck* Reaction C. Stille Reaction D. Suzuki* Reaction E. Sonogashira Reaction F. Buchwald-Hartwig Reaction . Summary 2 . Reactions that form (usually) carbon-carbon bonds between complex fragments . Typically use a transition metal catalyst and an organometallic precursor . Most involve a “transmetallation step” . Transmetallation: Transfer of alkyl group from one metal to another . Typical trend: Can transfer from more electropositive to less electropositive metals 3 Pd Catalyst . General reaction scheme: R-X + R . R: Lacks a β hydrogen attached to an sp3 carbon. (Aryl/Benzyl/Vinyl/Allyl) . X: Typically Cl, Br, I, Otf . Regioselectivity and rates are determined by steric hindrance at the alkene CH CHR RCH~ RRC 2 > RRC > HCR > H2C H2C H2C HCR 4 Spessard and Meissler, Organometallic Chemistry . Palladium is in the 0 oxidation state in the active catalyst : PPh3 Pd Ph3P PPh3 e PdCl2 0 . The palladium can be reduced in situ : Pd PPh3 Pd(OAc)2 . Preferred solvent is DMF . Increases rate, lowers temp. to from 800 C : - n Bu N+ ( )4 + KHCO3 . Two catalytic cycles are possible depending on the reaction conditions 5 Spessard and Meissler, Organometallic Chemistry Oxidative Addition: Pd(0) Pd(+2) d10 d8 14 e 16 e 6 Insertion: Pd(+2) Pd(+2) d8 d8 16 e 16 e 7 Hydride elimination: Pd(+2) Pd(+2) d8 d8 16 e 16 e 8 Reductive elimination: Pd(+2) Pd(0) d8 d10 16 e 14 e 9 Example of an intramolecular Heck reaction 10 BINAP . By preventing β elimination from the new sp3 center, an asymmetric product may be formed . -

Palladium-Catalyzed Carbonylation and Arylation Reactions
To Erik, Frida, Oskar & Camilla List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Sävmarker, J., Lindh, J., Nilsson, P. Deoxybenzoins from Stille Carbonylative Cross-Couplings using Molybdenum Hexa- carbonyl. Tetrahedron Lett. 2010, 51, 6886-6889. II Odell, L., Sävmarker, J., Larhed, M. Microwave-Promoted Aminocarbonylation of Aryl Triflates using Mo(CO)6 as a Solid CO Source. Tetrahedron Lett. 2008, 49, 6115-6118. III Lindh, J., Sävmarker, J., Nilsson, P., Sjöberg, P. J. R., Larhed, M. Synthesis of Styrenes by Palladium(II)-Catalyzed Vinylation of Arylboronic Acids and Aryltrifluoroborates by Using Vinyl Acetate. Chem-Eur J. 2009, 15, 4630-4636. IV Sävmarker, J., Lindh, J., Nilsson, P., Sjöberg, P. J. R., Larhed, M. Oxidative Heck Reactions Using Aryltrifluoroborates and Aryl MIDA Boronates. Submitted. V Sävmarker, J., Rydfjord, J., Gising, J., Odell, L., Larhed, M. Direct Palladium(II)-Catalyzed Synthesis of Arylamidines from Aryltrifluoroborates. Submitted. VI Behrends, M., Sävmarker, J., Sjöberg, P. J. R., Larhed, M. Microwave-Assisted Palladium(II)-Catalyzed Synthesis Of Aryl Ketones from Aryl Sulfinates and Direct ESI-MS Studies Thereof. ACS Catal. 2011, 1, 1455-1459. Reprints were made with permission from the respective publishers. Related Publications and Manuscript Not Included in this Thesis Trejos, A., Sävmarker, J., Schlummer, S., Datta, G. K., Nilsson, P., Larhed, M. Stereoselective Heck Arylation of a Functionalized Cyclopentenyl Ether using (S)-N-Methyl-Pyrrolidine as the Stereochemical Controller. Tetrahed- ron 2008, 64, 8746-8751. Andaloussi, M., Lindh, J. Sävmarker, J., Sjöberg, P. J. R., Larhed, M. -

1 Stille Polycondensation: a Versatile Synthetic Approach to Functional Polymers
1 1 Stille Polycondensation: A Versatile Synthetic Approach to Functional Polymers Tianyue Zheng, Alexander M. Schneider, and Luping Yu 1.1 Introduction The development of functional polymers is a very active research field that covers every aspects of our lives and has had huge impact on human society due to their applications in many cutting-edge technologies, such as energy conversion and storage, electronic devices, biotechnology, and health care, to name a few [1]. Scientists from different disciplines have invented numerous new materials for those purposes. Integral to these efforts is the development of efficient, versatile, and scalable synthesis techniques, which in turn enable the development of new functional materials. Thus, new synthetic methodologies are always a critical research topic that is actively pursued. A large number of recent advances can be cited to support this view, such as ring-opening metathesis polymerization (ROMP), atom transfer radical polymerization (ATRP, a type of “living” radical polymerization), and controlled Ziegler–Natta polymerization [2, 3]. Most recently, polycondensations based on transition metal-catalyzed CC bond formation reactions have emerged as important methodologies for synthesis of electro-optic materials containing large systems. These reactions include Stille, Suzuki, Negishi, Heck, and so on [4–7]. The Stille reaction is one of the best methods for the synthesis of organic functional materials due to its excellent compatibility with various functional groups and high reaction yield. The most attractive application of the Stille coupling reaction is in the synthesis of conjugated, polyaromatic semiconducting materials, which are an important class of materials for organic electronics. These materials exhibit good solubility in various sol- vents, which allows them to be fabricated into devices using inexpensive solution-phase printing techniques [8]. -

Stille Coupling of Α-Acyloxybenzylstannanes with Acid Chlorides
Stille Coupling of α-Acyloxybenzylstannanes with Acid Chlorides by Junhui Xu A thesis presented to the University of Waterloo in fulfillment of the thesis requirements for the degree of Master of Science in Chemistry Waterloo, Ontario, Canada, 2016 © Junhui Xu 2016 I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Two widely-accepted models (open- and cyclic- transmetalation) are applied to explain the stereochemical outcome of the Stille coupling with organotin compounds containing an sp3 chiral center. However, it is still not possible to predict the stereochemical outcome of this type of Stille coupling before the reaction is conducted. To have a better understanding of the detailed mechanism involved, the Stille couplings of different α-acyloxybenzylstannanes with different acid chlorides were studied in this project. The effects of several factors of both the yield and the stereochemical outcome of the Stille reaction containing an α-acyloxybenzylstannane have been studied. These factors were the protecting group on the organotin nucleophile, ligand, solvent and the substituent on the electrophile. It was found that both a bulky protecting group and ligand with mild σ-donicity could give good yield, while adding any substituent on the electrophile always lowered the yield of reaction. Retention of configuration was observed in the Stille coupling reaction containing an enantiomerically enriched α-alcyloxybenzylstannane. Potential racemization was found after the reaction. Adding a base or doubling the amount of CuCN successfully achieved retention of configuration with > 90% enantiospecificity (e.s.) regardless of the nature of the protecting group, substituent on the electrophile and phosphine ligand. -

Alkynes As Stille Reaction Pseudohalides: Gold- and Palladium-Cocatalyzed Synthesis of Tri- and Tetra-Substituted Olefins
Alkynes as Stille Reaction Pseudohalides: Gold- and Palladium-Cocatalyzed Synthesis of Tri- and Tetra-Substituted Olefins R 2 R2 Au(I)/Pd(0) Bu SnR R3 R2 (II)Pd Au 3 3 CO R Bu3Sn CO2R1 CO2R1 2 1 Yili Shi, Sonja M. Peterson, Walter W. Haberaecker III, and Suzanne A. Blum J. Am. Chem. Soc., 130 (7), 2168 -2169, 2008 Current Literature Chenbo Wang @ Wipf Group 2/16/2008 Chenbo Wang @ Wipf Group 1 2/16/2008 Carbometallation of Alkynes -General Concepts R R R R 1 2 R4 1 2 syn addition R M R R R3M 3 3 4 R1 R2 R M R R 1 R4 1 4 anti addition R3 R2 R3 R2 • A powerful way to make stereospecifically substituted alkenes. • M can be Cu, Al, Zr, Ti, Zn, B, Mg, Li or a combination of two. • Syn or anti selectivity depends on the nature of substrate, reagent and reaction conditions. • Regioselectivity depends on the polarity of the triple bond, steric factor and complexation by the functional groups of the alkyne. • Polyaddtitions: the product may add to the starting alkyne, resulting polymerization. For a review, see: Normant, J. F.; Alexakis, A. Synthesis 1981, 841. Chenbo Wang @ Wipf Group 2 2/16/2008 Carbometallation of Alkynes -Organocopper Reagents •Unfunctionalized Terminal alkynes: Markownikov rule R R1 1 + •MgX2 R Cu R2Cu•MgX2 2 •Functionalized alkynes: directed carbometallation RCu•MHal (CH2)n (CH2)n X X R Cu n = 0, 1, 2, 3 - X = halogen, SR, OR, O ,NR2,OAc Chenbo Wang @ Wipf Group 3 2/16/2008 Synthetic Use of Alkenylcopper(I) Derivatives R1 R1 R2 CO2H R1 R2 R3 R2 I O R3COCl I CO2 2 R R 1 R X R 1 3 1 R3MCl M = Sn, Si, P, S R R R MR 2 3 R Cu 2 3 O 2 O R3 R3 R4 R3CHO R1 OH R1 R2 R1 R3 R2 R O R R 3 2 3 R HO 4 Chenbo Wang @ Wipf Group 4 2/16/2008 Carbometallation of Alkynes -Organoaluminum Reagents R1 R CO H 2 R1 R I BuLi then CO2 I2 O R1 R cat Cp2ZrCl2 R1 OH 1 R + 3 R R AlR2 R R3Al 3 BuLi then R3X BuLi then R1 R3CHO R R3 R1 R R3 HO •Complementary to the organocopper reagents Negishi, E. -

The Stille Reaction Chem 115
Myers The Stille Reaction Chem 115 Recent Reviews: • Oxidative addition initally gives a cis complex that can rapidly isomerize to the trans isomer: Williams, R. Org. Synth. 2011, 88, 197–201. Selig, R.; Schollmeyer, D.; Albrecht, W.; Laufer, S. Tetrahedron 2011, 67, 9204–9213. R L PdL2 fast Tietze, L. F.; Dufert, A. Pure Appl. Chem., 2010, 82, 1375–1392. R–I L Pd I R Pd I L L Generalized Cross-Coupling: cis trans R–X R'–M catalyst R–R' M–X Casado, A. L.; Espinet, P. Organometallics 1998, 17, 954–959. • !-hydride elimination can be a serious side reaction within alkyl palladium intermediates. This typically requires a syn coplanar alignment of hydride and palladium: Typically: • R and R' are sp2–hybridized • M = Sn, B, Zr, Zn • X = I, OSO2CF3, Br, Cl • catalyst = Pd (sometimes Ni) H Pd(II)L2X + HPd(II)L2X Mechanism: • A specific example: • Oxidative-addition and reductive-elimination steps occur with retention of configuration for 2 Pd catalyst sp -hybridized substrates. p-Tol–Br + n-Bu3Sn–Ph p-Tol–Ph + n-Bu3Sn–Br Pd(II) • Transmetalation is proposed to be the rate-determining step with most substrates. • Relative order of ligand transfer from Sn: p-Tol–Ph Pd(0)Ln p-Tol–Br alkynyl > alkenyl > aryl > allyl = benzyl > "-alkoxyalkyl > alkyl reductive elimination oxidative addition • Electron-rich and sterically hindered aryl halides undergo slower oxidative addition and are often poor substrates as a result. p-Tol–Pd(II)Lm–Ph p-Tol–Pd(II)Lm–Br • Electron-poor stannanes undergo slower transmetallation and are often poor substrates as n-Bu Sn–Br n-Bu Sn–Ph 3 3 a result. -
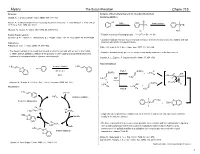
The Suzuki Reaction Chem 115 Reviews: Analysis of Elementary Steps in the Reaction Mechanism Suzuki, A
Myers The Suzuki Reaction Chem 115 Reviews: Analysis of Elementary Steps in the Reaction Mechanism Suzuki, A. J. Organometallic Chem. 1999, 576, 147–168. Oxidative Addition Br L Suzuki, A. In Metal-catalyzed Cross-coupling Reactions, Diederich, F., and Stang, P. J., Eds.; Wiley- Br 0 Pd Ln isomerization VCH: New York, 1998, pp. 49-97. Pd L PdII Br L L Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457-2483. cis trans B-Alkyl Suzuki reaction: • Relative reactivity of leaving groups: I – > OTf – > Br – >> Cl –. Chemler, S. R.; Trauner, D.; Danishefsky, S. J. Angew. Chem., Int. Ed. Engl. 2001, 40, 4544–4568. • Oxidative addition is known to proceed with retention of stereochemistry with vinyl halides and with Solid phase: inversion with allylic or benzylic halides. Franzén, R. Can. J. Chem. 2000, 78, 957–962. Stille, J. K.; Lau, K. S. Y. Acc. Chem. Res. 1977, 10, 434–442. • The Suzuki reaction is the coupling of an aryl or vinyl boronic acid with an aryl or vinyl halide • Oxidative addition intially gives a cis complex that rapidly isomerizes to its trans isomer. or triflate using a palladium catalyst. It is a powerful cross-coupling method that allows for the synthesis of conjugated olefins, styrenes, and biphenyls: Casado, A. L.; Espinet, P. Organometallics 1998, 17, 954–959. Transmetallation n-Bu benzene/NaOEt n-Bu O B + Br – O 80 ˚C, 4 h Path A B(OR)3 X B(OR)2 + L2Pd Ar H OR 98% n-Bu n-Bu B OR n-Bu RO– H + Miyaura, N.; Suzuki, A.