Lack of Transferrin Receptor 1 in the Heart Causes Lethal Cardiomyopathy And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Progressive Increase in Mtdna 3243A>G Heteroplasmy Causes Abrupt
Progressive increase in mtDNA 3243A>G PNAS PLUS heteroplasmy causes abrupt transcriptional reprogramming Martin Picarda, Jiangwen Zhangb, Saege Hancockc, Olga Derbenevaa, Ryan Golhard, Pawel Golike, Sean O’Hearnf, Shawn Levyg, Prasanth Potluria, Maria Lvovaa, Antonio Davilaa, Chun Shi Lina, Juan Carlos Perinh, Eric F. Rappaporth, Hakon Hakonarsonc, Ian A. Trouncei, Vincent Procaccioj, and Douglas C. Wallacea,1 aCenter for Mitochondrial and Epigenomic Medicine, Children’s Hospital of Philadelphia and the Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA 19104; bSchool of Biological Sciences, The University of Hong Kong, Hong Kong, People’s Republic of China; cTrovagene, San Diego, CA 92130; dCenter for Applied Genomics, Division of Genetics, Department of Pediatrics, and hNucleic Acid/Protein Research Core Facility, Children’s Hospital of Philadelphia, Philadelphia, PA 19104; eInstitute of Genetics and Biotechnology, Warsaw University, 00-927, Warsaw, Poland; fMorton Mower Central Research Laboratory, Sinai Hospital of Baltimore, Baltimore, MD 21215; gGenomics Sevices Laboratory, HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806; iCentre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, East Melbourne, VIC 3002, Australia; and jDepartment of Biochemistry and Genetics, National Center for Neurodegenerative and Mitochondrial Diseases, Centre Hospitalier Universitaire d’Angers, 49933 Angers, France Contributed by Douglas C. Wallace, August 1, 2014 (sent for review May -

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

Pharmacological Inhibition of Tumor Anabolism and Host Catabolism As A
www.nature.com/scientificreports OPEN Pharmacological inhibition of tumor anabolism and host catabolism as a cancer therapy Alejandro Schcolnik‑Cabrera1,2, Alma Chavez‑Blanco1, Guadalupe Dominguez‑Gomez1, Mandy Juarez1, Ariana Vargas‑Castillo3, Rafael Isaac Ponce‑Toledo4, Donna Lai5, Sheng Hua5, Armando R. Tovar3, Nimbe Torres3, Delia Perez‑Montiel6, Jose Diaz‑Chavez1 & Alfonso Duenas‑Gonzalez1,7* The malignant energetic demands are satisfed through glycolysis, glutaminolysis and de novo synthesis of fatty acids, while the host curses with a state of catabolism and systemic infammation. The concurrent inhibition of both, tumor anabolism and host catabolism, and their efect upon tumor growth and whole animal metabolism, have not been evaluated. We aimed to evaluate in colon cancer cells a combination of six agents directed to block the tumor anabolism (orlistat + lonidamine + DON) and the host catabolism (growth hormone + insulin + indomethacin). Treatment reduced cellular viability, clonogenic capacity and cell cycle progression. These efects were associated with decreased glycolysis and oxidative phosphorylation, leading to a quiescent energetic phenotype, and with an aberrant transcriptomic landscape showing dysregulation in multiple metabolic pathways. The in vivo evaluation revealed a signifcant tumor volume inhibition, without damage to normal tissues. The six‑drug combination preserved lean tissue and decreased fat loss, while the energy expenditure got decreased. Finally, a reduction in gene expression associated with thermogenesis was observed. Our fndings demonstrate that the simultaneous use of this six‑drug combination has anticancer efects by inducing a quiescent energetic phenotype of cultured cancer cells. Besides, the treatment is well‑ tolerated in mice and reduces whole animal energetic expenditure and fat loss. -

Robert Shine, Peter Dwyer, Lyndsay Olson, Jennifer Truong, Matthew Goddeeris, Effie Tozzo, Eric Bell Mitobridge, Inc
Poster Title Here Mitochondrial Deficiency in Primary Muscle Cells from Mdx Mice Robert Shine, Peter Dwyer, Lyndsay Olson, Jennifer Truong, Matthew Goddeeris, Effie Tozzo, Eric Bell Mitobridge, Inc. Cambridge, MA 02138 Abstract Results Results Duchenne muscular dystrophy (DMD) is a recessive, fatal X-linked disease that is characterized M ito c h o n d ria l c o n trib u te d A T P is re d u c e d Mdx myoblasts have decreased expression by progressive skeletal muscle wasting due to a loss of function in dystrophin, a protein that is of OXPHOS complexes part of a complex that bridges the cytoskeleton and extracellular matrix. The mdx mouse, an in m d x m y o b la s ts a n d m y o tu b e s T animal model for DMD, has a point mutation in the dystrophin gene that results in a loss of 1 .5 2.0 W l function. This study uses primary muscle satellite cell derived myoblasts and myotubes to W T a WT m determine differences in mitochondrial biology between the mdx mice and wild type (WT) control s M D X o 1.5 a r MDX mice. Compared to cells isolated from WT mice, mdx cells have reductions in mitochondrial 1 .0 **** f B bioenergetics. Moreover, mdx cells have reduced levels of mitochondria which may partially *** e T g 1.0 explain the reduction in bioenergetics. Interestingly, the mitochondrial phenotype is apparent **** ** n * * W a * * * before dystrophin protein is increased during myogenesis. * * * 0 .5 h 0.5 * d l C o d l F 0.0 o 1 3 2 6 1 a 1 a b B 0 .0 F t v Materials and Analysis a 5 P X D C H H y 5 l l F X T N R O D a M in a M in D C s s P n n U A c c O S C C a 0 a 0 S y y T B 0 B 0 D WT and mdx myoblast isolation and culture: Quadricep and gastrocnemius muscles from a C 1 m 1 m Q A o o N single mouse were pooled and subjected to a mechanical/collagenase digestion. -

Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: an Update
International Journal of Molecular Sciences Review Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update Philippe Icard 1,2,3,*, Antoine Coquerel 1,4, Zherui Wu 5 , Joseph Gligorov 6, David Fuks 7, Ludovic Fournel 3,8, Hubert Lincet 9,10 and Luca Simula 11 1 Medical School, Université Caen Normandie, CHU de Caen, 14000 Caen, France; [email protected] 2 UNICAEN, INSERM U1086 Interdisciplinary Research Unit for Cancer Prevention and Treatment, Normandie Université, 14000 Caen, France 3 Service de Chirurgie Thoracique, Hôpital Cochin, Hôpitaux Universitaires Paris Centre, APHP, Paris-Descartes University, 75014 Paris, France; [email protected] 4 INSERM U1075, COMETE Mobilités: Attention, Orientation, Chronobiologie, Université Caen, 14000 Caen, France 5 School of Medicine, Shenzhen University, Shenzhen 518000, China; [email protected] 6 Oncology Department, Tenon Hospital, Pierre et Marie Curie University, 75020 Paris, France; [email protected] 7 Service de Chirurgie Digestive et Hépato-Biliaire, Hôpital Cochin, Hôpitaux Universitaires Paris Centre, APHP, Paris-Descartes University, 75014 Paris, France; [email protected] 8 Descartes Faculty of Medicine, University of Paris, Paris Center, 75006 Paris, France 9 INSERM U1052, CNRS UMR5286, Cancer Research Center of Lyon (CRCL), 69008 Lyon, France; [email protected] 10 ISPB, Faculté de Pharmacie, Université Lyon 1, 69373 Lyon, France 11 Department of Infection, Immunity and Inflammation, Institut Cochin, INSERM U1016, CNRS UMR8104, Citation: Icard, P.; Coquerel, A.; Wu, University of Paris, 75014 Paris, France; [email protected] Z.; Gligorov, J.; Fuks, D.; Fournel, L.; * Correspondence: [email protected] Lincet, H.; Simula, L. -

PIM2-Mediated Phosphorylation of Hexokinase 2 Is Critical for Tumor Growth and Paclitaxel Resistance in Breast Cancer
Oncogene (2018) 37:5997–6009 https://doi.org/10.1038/s41388-018-0386-x ARTICLE PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer 1 1 1 1 1 2 2 3 Tingting Yang ● Chune Ren ● Pengyun Qiao ● Xue Han ● Li Wang ● Shijun Lv ● Yonghong Sun ● Zhijun Liu ● 3 1 Yu Du ● Zhenhai Yu Received: 3 December 2017 / Revised: 30 May 2018 / Accepted: 31 May 2018 / Published online: 9 July 2018 © The Author(s) 2018. This article is published with open access Abstract Hexokinase-II (HK2) is a key enzyme involved in glycolysis, which is required for breast cancer progression. However, the underlying post-translational mechanisms of HK2 activity are poorly understood. Here, we showed that Proviral Insertion in Murine Lymphomas 2 (PIM2) directly bound to HK2 and phosphorylated HK2 on Thr473. Biochemical analyses demonstrated that phosphorylated HK2 Thr473 promoted its protein stability through the chaperone-mediated autophagy (CMA) pathway, and the levels of PIM2 and pThr473-HK2 proteins were positively correlated with each other in human breast cancer. Furthermore, phosphorylation of HK2 on Thr473 increased HK2 enzyme activity and glycolysis, and 1234567890();,: 1234567890();,: enhanced glucose starvation-induced autophagy. As a result, phosphorylated HK2 Thr473 promoted breast cancer cell growth in vitro and in vivo. Interestingly, PIM2 kinase inhibitor SMI-4a could abrogate the effects of phosphorylated HK2 Thr473 on paclitaxel resistance in vitro and in vivo. Taken together, our findings indicated that PIM2 was a novel regulator of HK2, and suggested a new strategy to treat breast cancer. Introduction ATP molecules. -

The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
236 3 Journal of M Fex et al. Mitochondria in β-cells 236:3 R145–R159 Endocrinology REVIEW The pathogenetic role of β-cell mitochondria in type 2 diabetes Malin Fex1, Lisa M Nicholas1, Neelanjan Vishnu1, Anya Medina1, Vladimir V Sharoyko1, David G Nicholls1, Peter Spégel1,2 and Hindrik Mulder1 1Department of Clinical Sciences in Malmö, Unit of Molecular Metabolism, Lund University Diabetes Centre, Clinical Research Center, Malmö University Hospital, Lund University, Malmö, Sweden 2Department of Chemistry, Center for Analysis and Synthesis, Lund University, Sweden Correspondence should be addressed to H Mulder: [email protected] Abstract Mitochondrial metabolism is a major determinant of insulin secretion from pancreatic Key Words β-cells. Type 2 diabetes evolves when β-cells fail to release appropriate amounts f TCA cycle of insulin in response to glucose. This results in hyperglycemia and metabolic f coupling signal dysregulation. Evidence has recently been mounting that mitochondrial dysfunction f oxidative phosphorylation plays an important role in these processes. Monogenic dysfunction of mitochondria is a f mitochondrial transcription rare condition but causes a type 2 diabetes-like syndrome owing to β-cell failure. Here, f genetic variation we describe novel advances in research on mitochondrial dysfunction in the β-cell in type 2 diabetes, with a focus on human studies. Relevant studies in animal and cell models of the disease are described. Transcriptional and translational regulation in mitochondria are particularly emphasized. The role of metabolic enzymes and pathways and their impact on β-cell function in type 2 diabetes pathophysiology are discussed. The role of genetic variation in mitochondrial function leading to type 2 diabetes is highlighted. -

Functional Characterization of Epilepsy Associated GABRG2 Mutations
Functional Characterization of Epilepsy Associated GABRG2 Mutations By Mengnan Tian Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in Partial Fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmacology August, 2012 Nashville, Tennessee Approved: Robert L. Macdonald, M.D., Ph.D. Ronald Emeson, Ph.D. Kevin Ess, M.D., Ph.D. Katherine T. Murray, M.D. Douglas P. Mortlock, Ph.D. To my grandparents, Yunbo Tian and Zhenhua Cao. ii Acknowledgements Throughout my years pursuing Ph.D. degree, I have always been grateful for the opportunity working with an amazing complement of scientists. I would like to express my sincerest gratitude to my thesis advisor Dr. Robert Macdonald. He is by far the best mentor that I could have ever wished. He has provided me with excellent guidance and support, and given me the confidence to develop into an independent scientist. He granted me unprecedented freedom to explore new scientific fields and implement novel research strategies and techniques. I greatly appreciate all his trust in me, and it has fostered my confidence in performing researches. He has shown me, by his example, what a good scientist and mentor should be. I would like to thank my colleagues and collaborators in Macdonald lab. I have been collaborating with Xuan Huang since her rotation and after she joined our lab in 2009. We have also become good friends. She has made important contributions to many parts of my thesis studies, and provided brilliant critiques to help me improve the research plan. I am also deeply indebted to Dr. -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -
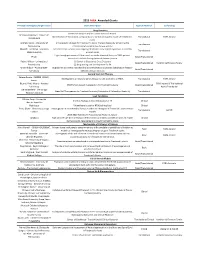
Grantsummaries 2014-2017 Jf
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's -

Network Organization of the Human Autophagy System
Vol 466 | 1 July 2010 | doi:10.1038/nature09204 ARTICLES Network organization of the human autophagy system Christian Behrends1, Mathew E. Sowa1, Steven P. Gygi2 & J. Wade Harper1 Autophagy, the process by which proteins and organelles are sequestered in autophagosomal vesicles and delivered to the lysosome/vacuole for degradation, provides a primary route for turnover of stable and defective cellular proteins. Defects in this system are linked with numerous human diseases. Although conserved protein kinase, lipid kinase and ubiquitin-like protein conjugation subnetworks controlling autophagosome formation and cargo recruitment have been defined, our understanding of the global organization of this system is limited. Here we report a proteomic analysis of the autophagy interaction network in human cells under conditions of ongoing (basal) autophagy, revealing a network of 751 interactions among 409 candidate interacting proteins with extensive connectivity among subnetworks. Many new autophagy interaction network components have roles in vesicle trafficking, protein or lipid phosphorylation and protein ubiquitination, and affect autophagosome number or flux when depleted by RNA interference. The six ATG8 orthologues in humans (MAP1LC3/GABARAP proteins) interact with a cohort of 67 proteins, with extensive binding partner overlap between family members, and frequent involvement of a conserved surface on ATG8 proteins known to interact with LC3-interacting regions in partner proteins. These studies provide a global view of the mammalian autophagy interaction landscape and a resource for mechanistic analysis of this critical protein homeostasis pathway. Protein homeostasis in eukaryotes is controlled by proteasomal turn- promote incorporation of Atg8p--PE into autophagosomes, allowing over of unstable proteins via the ubiquitin system and lysosomal turn- Atg8p to promote autophagosome closure and cargo recruitment. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation.