Simulating Coherent Multidimensional Spectroscopy of Nonadiabatic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rotational Spectroscopy
DAV University Jalandhar Department of Physics Study Material: PHY 512 Atomic & Molecular Spectroscopy M.Sc (Hons.) Physics 2nd sem Source : internet -http://www.phys.ubbcluj.ro/~dana.maniu/OS/BS_3.pdf BOOK: Fundamentals of Molecular Spectroscopy CN Banwell Mc Graw Hill Rotational spectroscopy - Involve transitions between rotational states of the molecules (gaseous state!) - Energy difference between rotational levels of molecules has the same order of magnitude with microwave energy - Rotational spectroscopy is called pure rotational spectroscopy, to distinguish it from roto-vibrational spectroscopy (the molecule changes its state of vibration and rotation simultaneously) and vibronic spectroscopy (the molecule changes its electronic state and vibrational state simultaneously) Molecules do not rotate around an arbitrary axis! Generally, the rotation is around the mass center of the molecule. The rotational axis must allow the conservation of M R α pα const kinetic angular momentum. α Rotational spectroscopy Rotation of diatomic molecule - Classical description Diatomic molecule = a system formed by 2 different masses linked together with a rigid connector (rigid rotor = the bond length is assumed to be fixed!). The system rotation around the mass center is equivalent with the rotation of a particle with the mass μ (reduced mass) around the center of mass. 2 2 2 2 m1m2 2 The moment of inertia: I miri m1r1 m2r2 R R i m1 m2 Moment of inertia (I) is the rotational equivalent of mass (m). Angular velocity () is the equivalent -

Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling
Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling Feng Xie,1 Floyd E. Dowell,2,3 and Xiuzhi S. Sun1 ABSTRACT Cereal Chem. 81(2):249–254 Starch, protein, and temperature effects on bread staling were inves- retrograded amylose-lipid complex was limited. Two important wave- tigated using visible and near-infrared spectroscopy (NIRS) and differ- lengths, 550 nm and 1,465 nm, were key for NIRS to successfully classify ential scanning calorimetry (DSC). Bread staling was mainly due to the starch-starch (SS) and starch-protein (SP) bread based on different amylopectin retrogradation. NIRS measured amylopectin retrogradation colors and protein contents in SS and SP. Low temperature dramatically accurately in different batches. Three important wavelengths, 970 nm, accelerated the amylopectin retrogradation process. Protein retarded bread 1,155 nm, and 1,395 nm, were associated with amylopectin retrogra- staling, but not as much as temperature. The starch and protein interaction dation. NIRS followed moisture and starch structure changes when was less important than the starch retrogradation. Protein hindered the amylopectin retrograded. The amylose-lipid complex changed little from bread staling process mainly by diluting starch and retarding starch retro- one day after baking. The capability of NIRS to measure changes in the gradation. Bread staling is a complex process that occurs during bread and starch (Osborne and Douglas 1981; Davies and Miller 1988; storage. It is a progressive deterioration of qualities such as taste, Millar et al 1996). Therefore, NIRS has the potential to provide fun- firmness, etc. -

Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification
Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification Chad Spreadbury, Regina Rodriguez, and David Mazyck College of Engineering, University of Florida Activated carbon (AC) is a proven effective adsorbent of contaminants in the air and water phases. This effectiveness is due to the large surface area of AC which hosts functional groups. Particularly, oxygen functional groups (e.g. carboxyls, lactones, phenols, carbonyls) have been noted as important in mercury removal. Hence, determining the quantities of these groups is crucial when AC is used for this purpose. Currently, Boehm titration is the most common method for determining the number of these functional groups. However, this test method is susceptible to high user error and require a lengthy procedure and run time. On the other hand, Fourier transform infrared (FTIR) spectroscopy excels at analyzing the functionality of an AC surface. This study investigates whether or not a correlation between Boehm titration and FTIR can be determined using quantitative and qualitative assessment. The results indicate that while using the current methodology for Boehm titrations and FTIR analysis, there is no clear correlation. However, this study does not rule out that a correlation may indeed exist between these test methods that can be elucidated with improved methodology that accounts for the unique physical and chemical characteristics of carbon. INTRODUCTION mercury (Hg0) to create oxidized mercury (Hg2+), which then undergoes chemisorption with the AC surface. ctivated carbon is used globally as an effective Boehm titrations and Fourier transform infrared (FTIR) adsorbent for removing contaminants like mercury spectroscopy are two common testing procedures for A(Hg) from air and water phases. -

Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-Typ
Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-type Cheese Split Defects THESIS Presented in Partial Fulfillment of the Requirements for the Master of Science Degree in the Graduate School of The Ohio State University By Feifei Hu Graduate Program in Food Science and Technology The Ohio State University 2012 Master's Examination Committee: Dr. W. James Harper, Advisor Dr. Luis E. Rodriguez-Saona Dr. Michael E. Mangino Copyright Feifei Hu 2012 Abstract Splits, slits and cracks are a continuing problem in the US Swiss Cheese industry and cause downgrading of the cheese, bringing economic loss. The defects generally occur during secondary fermentation in cold storage, which is known to be an issue caused by many factors. Many chemical and physical changes can be indicators to help predicting the defects. Various techniques have been applied to test different compounds in cheese. HPLC is one of the reference methods which have been applied in cheese chemistry for the past decades. Its application involves in the qualification and quantification of organic acids, amino acids and peptides, and sugars in dairy products. However, limited research has been conducted on a rapid and easy testing regimen to investigate split defects in Swiss-type cheese. For over fifty years, researchers have investigated the application of Fourier Transform Infrared (FTIR) Spectroscopy in various food samples, including cheeses and other dairy products because FTIR spectroscopy gives fast, simple and comprehensive detection of chemical compounds. There is great potential for applying FTIR spectroscopy in developing a rapid test method to detect and predict the occurrence of split defects. -

UC Santa Barbara Dissertation Template
UNIVERSITY OF CALIFORNIA Santa Barbara Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry by Jacob Alan Berenbeim Committee in charge: Professor Mattanjah de Vries, Chair Professor Steve Buratto Professor Michael Gordon Professor Martin Moskovits December 2017 The dissertation of Jacob Alan Berenbeim is approved. ____________________________________________ Steve Buratto ____________________________________________ Michael Gordon ____________________________________________ Martin Moskovits ____________________________________________ Mattanjah de Vries, Committee Chair October 2017 Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes Copyright © 2017 by Jacob Alan Berenbeim iii ACKNOWLEDGEMENTS To my wife Amy thank you for your endless support and for inspiring me to match your own relentless drive towards reaching our goals. To my parents and my brothers Eli and Gabe thank you for your love and visits to Santa Barbara, CA. To my advisor Mattanjah and my lab mates thank you for the incredible opportunity to share ideas and play puppets with the fabric of space. And to my cat Lola, you’re a good cat. iv VITA OF JACOB ALAN BERENBEIM October 2017 EDUCATION University of California, Santa Barbara CA Fall 2017 PhD, Physical Chemistry Advisor: Prof. Mattanjah S. de Vries University of Puget Sound, Tacoma WA 2009 BS, Chemistry Advisor: Prof. Daniel Burgard LABORATORY TECHNIQUES Photophysics by UV/VIS and IR pulsed laser spectroscopy, optical alignment, oa-TOF mass spectrometry (multiphoton ionization, MALDI, ESI+), molecular beam high vacuum apparatus, high voltage electronics, molecular computational modeling with Gaussian, data acquisition with LabView, and data manipulation with Mathematica and Origin RESEARCH EXPERIENCE Graduate Student Researcher 2012-2017 • Time dependent (transient) photo relaxation of organic molecules, including PAHs and aromatic biological molecules. -

Tuesday Morning, October 16, 2007
Tuesday Morning, October 16, 2007 Electronic Materials and Processing 9:20am EM-TuM5 Real-time Conductivity Analysis through Single- Molecule Electrical Junctions, J.-S. Na, J. Ayres, K.L. Chandra, C.B. Room: 612 - Session EM-TuM Gorman, G.N. Parsons, North Carolina State University We have recently developed a molecular electronic characterization test-bed Molecular Electronics that utilizes a symmetric pair of gold nanoparticles, 40 nm in diameter, joined together by a single or small group of conjugated oligomeric phenylene ethynylene (OPE) molecules. These Moderator: I. Hill, Dalhousie University nanoparticle/molecule/nanoparticle structures are subsequently assembled between nanoscale test electrodes to enable current through the molecule to 8:00am EM-TuM1 Dynamics of Molecular Switch Molecules Imaged be characterized. At low voltage (< ±1.5 V) the observed current is by Alternating Current Scanning Tunneling Microscopy, A.M. Moore, consistent with common non-resonant tunneling (i.e., I vs V is independent P.S. Weiss, The Pennsylvania State University of temperature between 80 and 300 K). This molecular analysis approach is We have studied oligo(phenylene-ethylene) (OPE) molecules as candidates unique because it enables the stability of the molecular conductance to be for molecular electronic switches. We previously determined the switching observed and characterized over extended periods (several weeks so far) mechanism to rely on hybridization changes between the substrate and after fabrication. Conductance through single molecule junctions was molecule. Here, we have determined which molecules will be more or less monitored in real-time during several process sequences, including active in our samples using a custom-built alternating current scanning dielectrophoretic directed self assembly and post-assembly modification. -

Raman Studies of Mass-Selected Metal Clusters
RAMAN STUDIES OF MASS-SELECTED METAL CLUSTERS Kenneth Andrew Bosnick A Thesis su bmitted in conformity with the requirements for the degree of Doctor of Philosophy in Experimental Physical Chemistry Graduate Department of Chemistry University of Toronto @ Copyright by Kenneth Andrew Bosnick 2000 National Library Bibliothèque nationale 1*1 of Canada du Canada Acquisitions and Acquisitions et Bibliographie Services services bibliographiques 395 Wellington Street 395. rue Wellington OîtawaON KlAW OnawaN KlAW Canada canada The author has granted a non- L'auteur a accordé une licence non exclusive licence aliowuig the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sel1 reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la fome de microfiche/film, de reproduction sur papier ou sur format électronique. The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des exîraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation. Sputtering a metal target under high vacuum conditions with 15 mA of 25 keV Ar' ions produces cationic metal clusters, which are extracted and collimated into a beam using standard ion optics. A particular cluster nuclearity is selected from the beam by a Wein filter, CO-depositedon an aluminum paddle with a matrix gas (Ar or CO) at cryogenic temperatures, and nectralized. -

Modeling of the Time-Resolved Vibronic Spectra of Polyatomic Molecules: the Formulation of the Problem and Analysis of Kinetic Equations S
Optics and Spectroscopy, Vol. 90, No. 2, 2001, pp. 199–206. Translated from Optika i Spektroskopiya, Vol. 90, No. 2, 2001, pp. 237–245. Original Russian Text Copyright © 2001 by Astakhov, Baranov. MOLECULAR SPECTROSCOPY Modeling of the Time-Resolved Vibronic Spectra of Polyatomic Molecules: The Formulation of the Problem and Analysis of Kinetic Equations S. A. Astakhov and V. I. Baranov Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, Moscow, 117975 Russia e-mail: [email protected] Received March 28, 2000 Abstract—A semiempirical parametric method is proposed for modeling three-dimensional (time-resolved) vibronic spectra of polyatomic molecules. The method is based on the use of the fragment approach in the for- mation of molecular models for excited electronic states and parametrization of these molecular fragments by modeling conventional (one-dimensional) absorption and fluorescence spectra of polyatomic molecules. All matrix elements that are required for calculations of the spectra can be found by the methods developed. The time dependences of the populations of a great number (>103) of vibronic levels can be most conveniently found by using the iterative numerical method of integration of kinetic equations. Convenient numerical algorithms and specialized software for PC are developed. Computer experiments showed the possibility of the real-time modeling three-dimensional spectra of polyatomic molecules containing several tens of atoms. © 2001 MAIK “Nauka/Interperiodica”. INTRODUCTION tions [14, 15]). Within the framework of the parametric theory of vibronic spectra, calculation methods were Recently, considerable advances have been made in developed which allow one to construct the spectral the experimental methods of molecular vibronic spec- ∆ν ≈ representation of the molecular model specified by a set troscopy. -
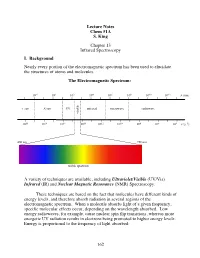
162 Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I
Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I. Background Nearly every portion of the electromagnetic spectrum has been used to elucidate the structures of atoms and molecules. The Electromagnetic Spectrum: 10!2 100 102 104 106 108 1010 1012 $ (nm) "!ray X-ray UV infrared microwave radiowave visible 1020 1018 1016 1014 1012 1010 108 106 104 # (s!1) 400 nm 750 nm ! visible spectrum A variety of techniques are available, including Ultraviolet/Visible (UV/Vis) Infrared (IR) and Nuclear Magnetic Resonance (NMR) Spectroscopy. These techniques are based on the fact that molecules have different kinds of energy levels, and therefore absorb radiation in several regions of the electromagnetic spectrum. When a molecule absorbs light of a given frequency, specific molecular effects occur, depending on the wavelength absorbed. Low energy radiowaves, for example, cause nuclear spin flip transitions, whereas more energetic UV radiation results in electrons being promoted to higher energy levels. Energy is proportional to the frequency of light absorbed: 162 Molecular effects associated with different regions of the EM spectrum: Wavelength (!) Energy/mole Molecular effects 10"10 meter gamma rays 106 kcal 10"8 meter X-rays 104 kcal ionization vaccum UV 102 kcal near UV electronic transitions 10"6 meter visible 10 kcal infrared 1 kcal molecular vibrations 10"4 meter (IR) 10"2 kcal 10"2 meter microwave rotational motion 10"4 kcal 0 10 meter radio 10"6 kcal nuclear spin transitions 2 10 meter II. IR Spectroscopy IR radiation causes groups of atoms to vibrate with respect to the bonds that connect them. -
Infrared and UV-Visible Time-Resolved Techniques for the Study of Tetrapyrrole-Based Proteins
Infrared and UV-visible time-resolved techniques for the study of tetrapyrrole-based proteins A thesis submitted to The University of Manchester for the degree of Doctor of Philosophy (PhD) in the Faculty of Life Sciences 2013 Henry John Russell Contents List of Tables .......................................................................................................................................................................... 5 List of Figures ........................................................................................................................................................................ 6 Abstract ...................................................................................................................................................................................10 Declaration ............................................................................................................................................................................11 Copyright Statement ........................................................................................................................................................11 Acknowledgements ..........................................................................................................................................................12 Abbreviations and Symbols ..........................................................................................................................................13 Preface and Thesis Structure .......................................................................................................................................16 -

Chapter 7 Raman Spectroscopy
Chapter 7 Raman Spectroscopy Course Code: SSCP 4473 Course Name: Spectroscopy & Materials Analysis Sib Krishna Ghoshal (PhD) Advanced Optical Materials Research Group Physics Department, Faculty of Science, UTM Essence of Raman Spectroscopy: Based on Inelastic Scattering Only 1 in 10,000,000 (0.00001%) photons are scattered inelastically Raman Effect is a 2-photon Scattering Process Presentation Menu Why to select Raman for materials analyses? Fundamental importance What is the process? When and Where I should do? Instrumentation Basic theoretical concepts Challenges Typical frequencies of Spectral analyses molecular vibrations range 12 Examples from less than 10 to approximately 1014 Hz. Conclusion What is Raman spectroscopy? A spectroscopic technique relies on inelastic scattering of monochromatic light (laser) in the visible, NIR, or NUV range. What is it for? For determining vibrational, rotational, and other low-frequency modes (fingerprints) in a system (molecule to bulk). Lysozyme is a protein found in How Raman spectra looks like? tears, saliva, and other secretions. Phe Trp C-H Trp Trp Lysozyme Graphene Trp Trp S-S C-S Phe Tyr His Phe Tyr Phe Trp Tyr Tyr Trp C-H Trp Phe Trp Mixed amino acid Phe Trp COOH Trp Tyr S-S What information C-S to extract ?? Tyr Need Analyses!! Human peptide and protein Phe 1750 1500 1250 1000 750 500 250 cm-1 Analyses of Samples Fingerprints Captured by Raman Spectra Characteristics Composition frequencies Number of bands Band Intensities Band position Stress/Strain Shifts in peak Band symmetry frequency State Band polarization Band width Polarization of Band shape peak Symmetry/ Orientation Spectrum: Raman Shift (cm-1) Vs. -

XXI European Conference on the Dynamics of Molecular Systems
XXI European Conference on the Dynamics of Molecular Systems Toledo (Spain) 11 - 16 September 2016 SPONSORS AND SUPPORTING ORGANIZATIONS INSTITUTIONAL SPONSORS SPONSORS WELCOME LETTER XXI European Conference on the Dynamics of Molecular Systems XXI European Conference on the Dynamics of Molecular Systems Dear participants in MOLEC 2016, Welcome to Toledo for the XXI edition of the MOLEC conference! The series of biennial MOLEC meetings started in 1976, when the first conference was held in Trento (Italy). Since that time the conference was held in Brandbjerg Hojskole (Denmark), Oxford (UK), Nijmegen (The Netherlands), Jerusalem (Israel), Aussois (France), Assisi (Italy), Bernkastel-Kues (Germany), Prague (Chech Republic), Salamanca (Spain), Nyborg Strand (Denmark), Bristol (UK), Jerusalem (Israel), Istanbul (Turkey), Nunspeet (The Netherlands), Trento (Italy), St. Petersburg (Russia), Curia (Portugal), Oxford (UK), Gothenburg (Sweden), and now in Toledo (Spain). The present conference aims at highlighting the most recent advances in molecular dynamics, providing the appropriate forum to discuss scientific achievements at the forefront of Chemistry and Physics. Following the tradition of MOLEC conferences, the scientific program focusses on both experimental and theoretical studies of molecular interactions, collisional dynamics, spectroscopy, and related fields. The present edition counts 107 participants from more than 20 countries, both European and non-European ones. The scientific program includes 46 talks and 67 poster contributions. We wish you a lively, fruitful, and successful conference in all respects. The time schedule allows time for informal discussions and exchange among participants. We also hope that participants there will have time to know and enjoy the beautiful and interesting city of Toledo. We would like to thank the International Scientific Committee for suggestions on the scientific program, and all the speakers for accepting the invitation to participate in MOLEC 2016.