Transcriptome Analysis Suggests a Compensatory Role of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inflammatory Stimuli Induce Acyl-Coa Thioesterase 7 and Remodeling of Phospholipids Containing Unsaturated Long (C20)-Acyl Chains in Macrophages
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2017/04/17/jlr.M076489.DC1 .html Inflammatory stimuli induce acyl-CoA thioesterase 7 and remodeling of phospholipids containing unsaturated long (C20)-acyl chains in macrophages Valerie Z. Wall,*,† Shelley Barnhart,* Farah Kramer,* Jenny E. Kanter,* Anuradha Vivekanandan-Giri,§ Subramaniam Pennathur,§ Chiara Bolego,** Jessica M. Ellis,§§,*** Miguel A. Gijón,††† Michael J. Wolfgang,*** and Karin E. Bornfeldt1,*,† Department of Medicine,* Division of Metabolism, Endocrinology and Nutrition, and Department of Pathology,† UW Medicine Diabetes Institute, University of Washington, Seattle, WA; Department of Internal Medicine,§ University of Michigan, Ann Arbor, MI; Department of Pharmaceutical and Pharmacological Sciences,** University of Padova, Padova, Italy; Department of Nutrition Science,§§ Purdue University, West Lafayette, IN; Department of Biological Chemistry,*** Johns Hopkins University School of Medicine, Baltimore, MD; and Department of Pharmacology,††† University of Downloaded from Colorado Denver, Aurora, CO Abstract Acyl-CoA thioesterase 7 (ACOT7) is an intracel- containing unsaturated long (C20)-acyl chains in macro- lular enzyme that converts acyl-CoAs to FFAs. ACOT7 is in- phages, and, although ACOT7 has preferential thioesterase duced by lipopolysaccharide (LPS); thus, we investigated activity toward these lipid species, loss of ACOT7 has no ma- www.jlr.org downstream effects of LPS-induced induction of ACOT7 jor detrimental effect on macrophage inflammatory pheno- and its role in inflammatory settings in myeloid cells. Enzy- types.—Wall, V. Z., S. Barnhart, F. Kramer, J. E. Kanter, matic thioesterase activity assays in WT and ACOT7-deficient A. Vivekanandan-Giri, S. Pennathur, C. Bolego, J. M. Ellis, macrophage lysates indicated that endogenous ACOT7 con- M. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Suppression of Fatty Acid Oxidation by Thioesterase Superfamily Member
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440732; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Suppression of Fatty Acid Oxidation by Thioesterase Superfamily Member 2 in Skeletal Muscle Promotes Hepatic Steatosis and Insulin Resistance Norihiro Imai1, Hayley T. Nicholls1, Michele Alves-Bezerra1, Yingxia Li1, Anna A. Ivanova2, Eric A. Ortlund2, and David E. Cohen1 1Division of Gastroenterology and Hepatology, Joan & Sanford I. Weill Department of Medicine, Weill Cornell Medical College, NY 10021 USA 2Department of Biochemistry, Emory University, Atlanta, GA 30322 USA Current addresses: Norihiro Imai - Department of Gastroenterology and Hepatology, Nagoya University School of Medicine, Aichi 4668560 Japan Michele Alves-Bezerra - Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030 USA bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440732; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Figure number: 8 Supplemental figure number: 10 Supplemental table number: 2 References: 48 Keywords: Hepatic steatosis, obesity, acyl-CoA thioesterase, fatty acid oxidation, insulin resistance Conflict of interest: The authors have declared that no conflict of interest exists. Author contributions: N.I.: designed research studies, conducted experiments, acquired data, analyzed data and wrote manuscript. H.T.N.: conducted experiments and analyzed data, M.A.B.: designed research studies and conducted experiments, Y.L.: acquired data, A.A.I.: conducted experiments and analyzed data, E.A.O.: analyzed data, D.E.C.: designed research studies, analyzed data and wrote manuscript. -

Structural Basis for Recruitment of Tandem Hotdog Domains in Acyl-Coa Thioesterase 7 and Its Role in Inflammation
Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation Jade K. Forwood*†‡, Anil S. Thakur*, Gregor Guncar*§¶, Mary Marfori*, Dmitri Mouradov*, Weining Meng*§, Jodie Robinson§ʈ, Thomas Huber*, Stuart Kellie*§ʈ, Jennifer L. Martin*§**, David A. Hume*§ʈ**††, and Bostjan Kobe*‡§** *School of Molecular and Microbial Sciences, §Institute for Molecular Bioscience, ʈCooperative Research Centre for Chronic Inflammatory Diseases, and **Australian Research Council Special Research Centre for Functional and Applied Genomics, University of Queensland, Brisbane, Queensland 4072, Australia Edited by Gregory A. Petsko, Brandeis University, Waltham, MA, and approved May 10, 2007 (received for review February 2, 2007) Acyl-CoA thioesterases (Acots) catalyze the hydrolysis of fatty long-chain acyl-CoA substrates with fatty acid chains of 8–16 acyl-CoA to free fatty acid and CoA and thereby regulate lipid carbon atoms (C8–C16) (7, 13). Acot7 contains a pair of fused metabolism and cellular signaling. We present a comprehensive thioesterase domains that share Ϸ30% sequence identity, with each structural and functional characterization of mouse acyl-CoA thio- thioesterase domain predicted to have the hotdog fold structure esterase 7 (Acot7). Whereas prokaryotic homologues possess a with an ␣-helix sausage wrapped by a -sheet bun (14–16). single thioesterase domain, mammalian Acot7 contains a pair of Here, we present the crystal structures of the N- and C- domains in tandem. We determined the crystal structures of both terminal thioesterase domains (N- and C-domains) of mouse the N- and C-terminal domains of the mouse enzyme, and inferred Acot7, refined at 1.8 and 2.4 Å resolution, respectively. -

The Emerging Role of Acyl-Coa Thioesterases and Acyltransferases in Regulating Peroxisomal Lipid Metabolism☆
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1822 (2012) 1397–1410 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Review The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism☆ Mary C. Hunt a,⁎, Marina I. Siponen b,1, Stefan E.H. Alexson c a Dublin Institute of Technology, School of Biological Sciences, College of Sciences & Health, Kevin Street, Dublin 8, Ireland b Department of Medical Biochemistry and Biophysics, Structural Genomics Consortium, Karolinska Institutet, SE-171 77 Stockholm, Sweden c Karolinska Institutet, Department of Laboratory Medicine, Division of Clinical Chemistry, Karolinska University Hospital at Huddinge, SE-141 86 Stockholm, Sweden article info abstract Article history: The importance of peroxisomes in lipid metabolism is now well established and peroxisomes contain approx- Received 15 November 2011 imately 60 enzymes involved in these lipid metabolic pathways. Several acyl-CoA thioesterase enzymes Received in revised form 3 March 2012 (ACOTs) have been identified in peroxisomes that catalyze the hydrolysis of acyl-CoAs (short-, medium-, Accepted 16 March 2012 long- and very long-chain), bile acid-CoAs, and methyl branched-CoAs, to the free fatty acid and coenzyme Available online 23 March 2012 A. A number of acyltransferase enzymes, which are structurally and functionally related to ACOTs, have also been identified in peroxisomes, which conjugate (or amidate) bile acid-CoAs and acyl-CoAs to amino acids, Keywords: Acyl-CoA thioesterase resulting in the production of amidated bile acids and fatty acids. -

Loss of the E3 Ubiquitin Ligase MKRN1 Represses Diet-Induced Metabolic Syndrome Through AMPK Activation
ARTICLE DOI: 10.1038/s41467-018-05721-4 OPEN Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation Min-Sik Lee1, Hyun-Ji Han2, Su Yeon Han2, Il Young Kim3,4, Sehyun Chae5, Choong-Sil Lee2, Sung Eun Kim2, Seul Gi Yoon4, Jun-Won Park4, Jung-Hoon Kim2, Soyeon Shin2, Manhyung Jeong2, Aram Ko2, Ho-Young Lee6, Kyoung-Jin Oh 7, Yun-Hee Lee 8, Kwang-Hee Bae7, Seung-Hoi Koo9, Jea-woo Kim10, Je Kyung Seong3,4, Daehee Hwang5 & Jaewhan Song 2 1234567890():,; AMP-activated protein kinase (AMPK) plays a key role in controlling energy metabolism in response to physiological and nutritional status. Although AMPK activation has been pro- posed as a promising molecular target for treating obesity and its related comorbidities, the use of pharmacological AMPK activators has been met with contradictory therapeutic challenges. Here we show a regulatory mechanism for AMPK through its ubiquitination and degradation by the E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1). MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation due to AMPK stabilisation and activation. Accordingly, MKRN1-null mice show chronic AMPK activation in both liver and adipose tissue, resulting in significant suppression of diet-induced metabolic syndrome. We demonstrate also its therapeutic effect by administering shRNA targeting MKRN1 into obese mice that reverses non-alcoholic fatty liver disease. We suggest that ubiquitin-dependent AMPK degradation represents a target therapeutic strategy for meta- bolic disorders. 1 Harvard Medical School, Boston Children’s Hospital, 3 Blackfan Circle CLS-16060.2, Boston, MA 02115, USA. 2 Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Republic of Korea. -

Role of Intramitochondrial Arachidonic Acid and Acyl-Coa Synthetase 4 in Angiotensin II-Regulated Aldosterone Synthesis in NCI-H295R Adrenocortical Cell Line Pablo G
GLUCOCORTICOIDS-CRH-ACTH-ADRENAL Role of Intramitochondrial Arachidonic Acid and Acyl-CoA Synthetase 4 in Angiotensin II-Regulated Aldosterone Synthesis in NCI-H295R Adrenocortical Cell Line Pablo G. Mele,* Alejandra Duarte,* Cristina Paz, Alessandro Capponi, and Ernesto J. Podestá Department of Biochemistry (P.G.M., A.D., C.P., E.J.P.), School of Medicine, University of Buenos Aires, Institute of Biomedical Investigations, UBA-CONICET, Buenos Aires, Argentina; and Division of Endocrinology (A.C.), University Hospital, Genève 14, Switzerland Although the role of arachidonic acid (AA) in angiotensin II (ANG II)- and potassium-stimulated steroid production in zona glomerulosa cells is well documented, the mechanism responsible for AA release is not fully described. In this study we evaluated the mechanism involved in the release of intramitochondrial AA and its role in the regulation of aldosterone synthesis by ANG II in glomerulosa cells. We show that ANG II and potassium induce the expression of acyl-coenzyme A (CoA) thioesterase 2 and acyl-CoA synthetase 4, two enzymes involved in intramitochondrial AA generation/export system well characterized in other steroidogenic systems. We demonstrate that mitochondrial ATP is required for AA generation/export system, steroid production, and steroid- ogenic acute regulatory protein induction. We also demonstrate the role of protein tyrosine phos- phatases regulating acyl-CoA synthetase 4 and steroidogenic acute regulatory protein induction, and hence ANG II-stimulated aldosterone synthesis. (Endocrinology 153: 3284–3294, 2012) teroid hormones, required for normal reproductive steroidogenic acute regulatory (StAR) protein, both mi- Sfunction and body homeostasis, are synthesized in ste- tochondrial proteins, are widely studied (4, 5). -
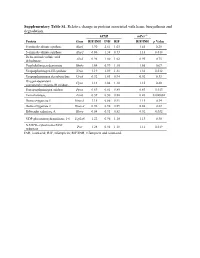
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

Organic & Biomolecular Chemistry
Organic & Biomolecular Chemistry Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/obc Page 1 of 7 Journal Name Organic & Biomolecular Chemistry Dynamic Article Links ► Cite this: DOI: 10.1039/c0xx00000x www.rsc.org/xxxxxx ARTICLE TYPE A study on the chiral inversion of mandelic acid in humans Maksims Yevglevskis, Catherine R. Bowskill, Chloe C. Y. Chan, Justin H.-J. Heng, Michael D. Threadgill, Timothy J. Woodman, and Matthew D. Lloyd* Received (in XXX, XXX) Xth XXXXXXXXX 20XX, Accepted Xth XXXXXXXXX 20XX 5 DOI: 10.1039/b000000x Manuscript Mandelic acid is a chiral metabolite of the industrial pollutant styrene and is used in chemical skin peels, as a urinary antiseptic and as a component of other medicines. -

Differential Regulation of Gene Expression by Cholesterol
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2016/05/25/jpet.116.233312.DC1 1521-0103/358/2/216–229$25.00 http://dx.doi.org/10.1124/jpet.116.233312 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 358:216–229, August 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics Differential Regulation of Gene Expression by Cholesterol Biosynthesis Inhibitors That Reduce (Pravastatin) or Enhance (Squalestatin 1) Nonsterol Isoprenoid Levels in Primary Cultured Mouse and Rat Hepatocytes s Elizabeth A. Rondini, Zofia Duniec-Dmuchowski, Daniela Cukovic, Alan A. Dombkowski, and Thomas A. Kocarek Institute of Environmental Health Sciences (E.A.R., Z.D.-D., T.A.K.), and Department of Pediatrics, Division of Clinical Pharmacology and Toxicology (D.C., A.A.D.), Wayne State University, Detroit, Michigan Downloaded from Received March 30, 2016; accepted May 24, 2016 ABSTRACT Squalene synthase inhibitors (SSIs), such as squalestatin 1 (SQ1), directly, 162 orthologs were found to be differentially coregu- reduce cholesterol biosynthesis but cause the accumulation of lated between the two treatments. Genes involved in cholesterol jpet.aspetjournals.org isoprenoids derived from farnesyl pyrophosphate (FPP), which and unsaturated fatty acid biosynthesis were up-regulated by can modulate the activity of nuclear receptors, including the both inhibitors, consistent with cholesterol depletion; however, constitutive androstane receptor (CAR), farnesoid X receptor, the extent of induction was greater in rat than in mouse hepa- and peroxisome proliferator-activated receptors (PPARs). In tocytes. SQ1 induced several orthologs associated with micro- comparison, 3-hydroxy-3-methylglutaryl-coenzyme A reduc- somal, peroxisomal, and mitochondrial fatty acid oxidation and tase inhibitors (e.g., pravastatin) inhibit production of both repressed orthologs involved in cell cycle regulation. -

ACOT1/2 (G-20): Sc-82473
SAN TA C RUZ BI OTEC HNOL OG Y, INC . ACOT1/2 (G-20): sc-82473 BACKGROUND APPLICATIONS Acyl-CoA thioesterases (ACOTs) are a group of enzymes that catalyze the hy- ACOT1/2 (G-20) is recommended for detection of ACOT1 and ACOT2 of drolysis of acyl-CoA to form coenzyme A (CoA) and a free fatty acid. Through human origin by Western Blotting (starting dilution 1:200, dilution range their catalytic activity, ACOTs are able to regulate the level of fatty acids and 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein acyl-CoAs within the cell. ACOT1 (acyl-CoA thioesterase 1, also known as (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution CTE1) and ACOT2 (acyl-CoA thioesterase 2, also known as PTE2) are members range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution of the ACOT family and exhibit different cellular localization, with ACOT1 range 1:30-1:3000); non cross-reactive with family member ACOT4. existing as a monomer in the cytoplasm and ACOT2 localized primarily to Molecular Weight of ACOT1: 46 kDa. mitochondria. Characteristic of most ACOT proteins, ACOT1 and ACOT2 cat - alyze the conversion of Palmitoyl-CoA and water to free CoA and palmitate, Molecular Weight of ACOT2: 53 kDa. a reaction that is important for the regulation of intercellular fatty acid levels. Positive Controls: HUV-EC-C whole cell lysate: sc-364180, MDA-MB-435S ACOT2 is expressed as multiple alternatively spliced isoforms and, like ACOT1, whole cell lysate: sc-364184 or HeLa whole cell lysate: sc-2200. -
ACYL-Coa THIOESTERASES- AUXILIARY ENZYMES in PEROXISOMAL LIPID METABOLISM
From the Department of Laboratory Medicine, Division of Clinical Chemistry Karolinska Institutet, Stockholm, Sweden ACYL-CoA THIOESTERASES- AUXILIARY ENZYMES IN PEROXISOMAL LIPID METABOLISM Maria A. K. Westin Stockholm 2007 Published and printed by Karolinska University Press Box 200, SE-171 77 Stockholm, Sweden © Maria A. K. Westin, 2007 ISBN 978-91-7357-241-5 Dedicated to the future reader ‘En enda droppe gör havet större’ -Anonym ABSTRACT Peroxisomes are small organelles essential for life, which carry out a variety of functions mostly related to lipid metabolism, including α- and β-oxidation as well as the first steps of plasmalogen biosynthesis. Peroxisomal β-oxidation metabolizes many different lipids including very long-chain fatty acids, dicarboxylic fatty acids, branched-chain fatty acids, eicosanoids and certain xenobiotic fatty acids. This PhD project began with the identification of four novel putative peroxisomal acyl- CoA thioesterase genes, named acyl-CoA thioesterase 3-6 (Acot3-Acot6). The acyl- CoA thioesterases are a group of enzymes that catalyze the hydrolysis of acyl-CoAs to free fatty acids and coenzyme A. The identification of these novel acyl-CoA thioesterases raised questions as to their function(s) and what the need is for so many peroxisomal acyl-CoA thioesterases. Answering these questions was therefore the aim of this project. Biochemical characterization showed that expression of all four enzymes is upregulated by peroxisome proliferators in a peroxisome proliferator- activated receptor α (PPARα) dependent manner, suggesting that they function in fatty acid metabolism. Further characterization revealed that ACOT3 and ACOT5 are a long- and a medium-chain acyl-CoA thioesterase, respectively, while ACOT4 and ACOT6 were shown to be specific enzymes catalyzing the hydrolysis of succinyl-CoA and phytanoyl-CoA/pristanoyl-CoA, respectively.