Temperature-Dependent Phase Behaviour of Tetrahydrofuran–Water
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Glossary Terms
Glossary Terms € 1584 5W6 5501 a 7181, 12203 5’UTR 8126 a-g Transformation 6938 6Q1 5500 r 7181 6W1 5501 b 7181 a 12202 b-b Transformation 6938 A 12202 d 7181 AAV 10815 Z 1584 Abandoned mines 6646 c 5499 Abiotic factor 148 f 5499 Abiotic 10139, 11375 f,b 5499 Abiotic stress 1, 10732 f,i, 5499 Ablation 2761 m 5499 ABR 1145 th 5499 Abscisic acid 9145 th,Carnot 5499 Absolute humidity 893 th,Otto 5499 Absorbed dose 3022, 4905, 8387, 8448, 8559, 11026 v 5499 Absorber 2349 Ф 12203 Absorber tube 9562 g 5499 Absorption, a(l) 8952 gb 5499 Absorption coefficient 309 abs lmax 5174 Absorption 309, 4774, 10139, 12293 em lmax 5174 Absorptivity or absorptance (a) 9449 μ1, First molecular weight moment 4617 Abstract community 3278 o 12203 Abuse 6098 ’ 5500 AC motor 11523 F 5174 AC 9432 Fem 5174 ACC 6449, 6951 r 12203 Acceleration method 9851 ra,i 5500 Acceptable limit 3515 s 12203 Access time 1854 t 5500 Accessible ecosystem 10796 y 12203 Accident 3515 1Q2 5500 Acclimation 3253, 7229 1W2 5501 Acclimatization 10732 2W3 5501 Accretion 2761 3 Phase boundary 8328 Accumulation 2761 3D Pose estimation 10590 Acetosyringone 2583 3Dpol 8126 Acid deposition 167 3W4 5501 Acid drainage 6665 3’UTR 8126 Acid neutralizing capacity (ANC) 167 4W5 5501 Acid (rock or mine) drainage 6646 12316 Glossary Terms Acidity constant 11912 Adverse effect 3620 Acidophile 6646 Adverse health effect 206 Acoustic power level (LW) 12275 AEM 372 ACPE 8123 AER 1426, 8112 Acquired immunodeficiency syndrome (AIDS) 4997, Aerobic 10139 11129 Aerodynamic diameter 167, 206 ACS 4957 Aerodynamic -

Membrane Phase Miscibility Gaps and Surface Constraints
SurfaceMembrane constraints phase miscibility and membrane gaps and phase surface miscibility constraints gaps W.A. Hamilton,1 L. Porcar1,2 and G.S. Smith3,1 1 Neutron Scattering Center, Oak Ridge National Laboratory 2 NIST Center for Neutron Research 3 LANSCE, Los Alamos National Laboratory Research supported by the US Department of Energy, Division of Materials Science 2nd American Conference on Neutron Scattering, College Park MD 6-10 June 2004 Self-assembled surfactant membrane phase: L3 “sponge” phase δ ~ 20-30Å bilayer membrane surfactant molecule d ~ 100 –1000 Å 3 “oily-salt” QuartzA surface very fluid aqueous phase over? wide dilution Potentially useful as a mixing stage in (membrane) protein crystallization for structural characterization and as template phaseOur for simply high porosityconstraining structures proximate (as surface per aerogels ... ) What does an isotropic bulk phase do in an anisotropic situation? Same in all directions - manifestly isotropic - so ... A nearby answer in the phase diagram … Generic membrane phase diagram region … A competition between curvature, topology and entropy e r Sponge - L u 3 t a v r ratio, salt d u I+L3 +… 3 hexanol c / c i s L3 n CPCl i r t or L3 + Lα biphasic - miscibility gap n cosurfactant i no single phase solution to competition here g n i s Lα Lamellar - L a α e AOT/brine r c Surfactant/ T ~1% ~50 % n i volume fraction φ dα Stacked lamellar phase would fit nicely against a constraining surface ... Near surface constraint contributes to F Expect local effect on phase transition and miscibility gap (Lα+L3 coexistence) Typical L3 and Lα SANS at highish volume fraction e.g. -

Miscibility in Solids
Name _________________________ Date ____________ Regents Chemistry Miscibility In Solids I. Introduction: Two substances in the same phases are miscible if they may be completely mixed (in liquids a meniscus would not appear). Substances are said to be immiscible if the will not mix and remain two distinct phases. A. Intro Activity: 1. Add 10 mL of olive oil to 10 mL of water. a) Please record any observations: ___________________________________________________ ___________________________________________________ b) Based upon your observations, indicate if the liquids are miscible or immiscible. Support your answer. ________________________________________________________ ________________________________________________________ 2. Now add 10 mL of grain alcohol to 10 mL of water. a) Please record any observations: ___________________________________________________ ___________________________________________________ b) Based upon your observations, indicate if the liquids are miscible or immiscible. Support your answer. ________________________________________________________ ________________________________________________________ Support for Cornell Center for Materials Research is provided through NSF Grant DMR-0079992 Copyright 2004 CCMR Educational Programs. All rights reserved. II. Observation Of Perthite A. Perthite Observation: With the hand specimens and hand lenses provided, list as many observations possible in the space below. _________________________________________________________ _________________________________________________________ -

Contact Melting and the Structure of Binary Eutectic Near the Eutectic Point
Contact melting and the structure of binary eutectic near the eutectic point Bystrenko O.V. 1,2 and Kartuzov V.V. 1 1 Frantsevich Institute for material sсience problems, Kiev, Ukraine, 2 Bogolyubov Institute for theoretical physics, Kiev, Ukraine Abstract. Computer simulations of contact melting and associated interfacial phenomena in binary eutectic systems were performed on the basis of the standard phase-field model with miscibility gap in solid state. It is shown that the model predicts the existence of equilibrium three-phase (solid-liquid-solid) states above the eutectic temperature, which suggest the explanation of the phenomenon of phase separation in liquid eutectic observed in experiments. The results of simulations provide the interpretation for the phenomena of contact melting and formation of diffusion zone observed in the experiments with binary metal-silicon systems. Key words: phase field, eutectic, diffusion zone, phase separation 1. Introduction. Phenomenon of contact (eutectic) melting (CM) is rather common for multicomponent systems and has important industrial implications [1], which motivate its experimental and theoretical studies. The aim of this work is a theoretical study of the properties of CM in binary eutectic systems and the interpretation of the results of recent experiments particularly focused on the investigation of the interfacial phenomena associated with CM [2, 3]. In these experiments, the binary systems consisting of metal Au, Al, Ag, and Cu particles (of 5 10-6 m size) placed on amorphous or crystalline -

Dihydrolevoglucosenone (Cyrene™), a Bio-Based Solvent for Liquid-Liquid Extraction Applications Thomas Brouwer, and Boelo Schuur ACS Sustainable Chem
Subscriber access provided by UNIV TWENTE Article Dihydrolevoglucosenone (Cyrene™), a Bio-based Solvent for Liquid-Liquid Extraction Applications Thomas Brouwer, and Boelo Schuur ACS Sustainable Chem. Eng., Just Accepted Manuscript • DOI: 10.1021/ acssuschemeng.0c04159 • Publication Date (Web): 31 Aug 2020 Downloaded from pubs.acs.org on September 16, 2020 Just Accepted “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts. is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 Published by American Chemical Society. -

An Effective Algorithm to Identify the Miscibility Gap in a Binary Substitutional Solution Phase
J. Min. Metall. Sect. B-Metall., 56 (2) B (2020) 183 - 191 Journal of Mining and Metallurgy, Section B: Metallurgy An effectIve AlgOrIthM tO IdentIfy the MIScIBIlIty gAp In A BInAry SuBStItutIOnAl SOlutIOn phASe t. fua, y. du b,*, Z.-S. Zheng a,*, y.-B. peng c, B. Jin b, y.-l. liu b, c.-f. du a, S.-h. liu b, c.-y. Shi b, J. Wang b a* School of Mathematics and Statistics, Central South University, Changsha, Hunan, China b State Key Laboratory of Powder Metallurgy, Central South University, Changsha, Hunan, China c College of Metallurgy and Materials Engineering, Hunan University of Technology, Zhuzhou, Hunan, China (Received 16 September 2019; accepted 18 May 2020) Abstract In the literature, no detailed description is reported about how to detect if a miscibility gap exists in terms of interaction parameters analytically. In this work, a method to determine the likelihood of the presence of a miscibility gap in a binary substitutional solution phase is proposed in terms of interaction parameters. The range of the last interaction parameter along with the former parameters is analyzed for a set of self-consistent parameters associated with the miscibility gap in assessment process. Furthermore, we deduce the first and second derivatives of Gibbs energy with respect to composition for a phase described with a sublattice model in a binary system. The Al-Zn and Al-In phase diagrams are computed by using a home-made code to verify the efficiency of these techniques. The method to detect the miscibility gap in terms of interaction parameters can be generalized to sublattice models. -

Phase Diagrams of Ternary -Conjugated Polymer Solutions For
polymers Article Phase Diagrams of Ternary π-Conjugated Polymer Solutions for Organic Photovoltaics Jung Yong Kim School of Chemical Engineering and Materials Science and Engineering, Jimma Institute of Technology, Jimma University, Post Office Box 378 Jimma, Ethiopia; [email protected] Abstract: Phase diagrams of ternary conjugated polymer solutions were constructed based on Flory-Huggins lattice theory with a constant interaction parameter. For this purpose, the poly(3- hexylthiophene-2,5-diyl) (P3HT) solution as a model system was investigated as a function of temperature, molecular weight (or chain length), solvent species, processing additives, and electron- accepting small molecules. Then, other high-performance conjugated polymers such as PTB7 and PffBT4T-2OD were also studied in the same vein of demixing processes. Herein, the liquid-liquid phase transition is processed through the nucleation and growth of the metastable phase or the spontaneous spinodal decomposition of the unstable phase. Resultantly, the versatile binodal, spinodal, tie line, and critical point were calculated depending on the Flory-Huggins interaction parameter as well as the relative molar volume of each component. These findings may pave the way to rationally understand the phase behavior of solvent-polymer-fullerene (or nonfullerene) systems at the interface of organic photovoltaics and molecular thermodynamics. Keywords: conjugated polymer; phase diagram; ternary; polymer solutions; polymer blends; Flory- Huggins theory; polymer solar cells; organic photovoltaics; organic electronics Citation: Kim, J.Y. Phase Diagrams of Ternary π-Conjugated Polymer 1. Introduction Solutions for Organic Photovoltaics. Polymers 2021, 13, 983. https:// Since Flory-Huggins lattice theory was conceived in 1942, it has been widely used be- doi.org/10.3390/polym13060983 cause of its capability of capturing the phase behavior of polymer solutions and blends [1–3]. -

Inside the Miscibility Gap Nanostructuring and Phase Transformations in Hard Nitride Coatings
Linköping Studies in Science and Technology Dissertation No. 1472 Inside The Miscibility Gap Nanostructuring and Phase Transformations in Hard Nitride Coatings Lars Johnson Thin Film Physics Division Department of Physics, Chemistry, and Biology (IFM) Linköping University SE-581 83 Linköping, Sweden Linköping 2012 © Lars Johnson Except for papers 1-3 © Elsevier B.V., used with permission. ISBN 978-91-7519-809-5 ISSN 0345-7524 Typeset using LATEX Printed by LiU-Tryck, Linköping 2012 II ABSTRACT This thesis is concerned with self-organization phenomena in hard and wear resistant transition-metal nitride coatings, both during growth and during post-deposition thermal annealing. The uniting physical principle in the studied systems is the immiscibility of their constituent parts, which leads, under certain conditions, to structural variations on the nanoscale. The study of such structures is challenging, and during this work atom probe to- mography (apt) was developed as a viable tool for their study. Ti0.33Al0.67N was observed to undergo spinodal decomposition upon annealing to 900 °C, by the use of apt in combination with electron microscopy. The addition of C to TiSiN was found to promote and refine the feather-like microstructure common in the system, with an ensuing decrease in thermal stability. An age-hardening of 36 % was measured in arc evaporated Zr0.44Al0.56N1.20, which was a nanocomposite of cubic, hexagonal, and amorphous phases. Magnetron sputtering of Zr0.64Al0.36N at 900 °C resulted in a self-organized and highly ordered growth of a two-dimensional two-phase labyrinthine structure of cubic ZrN and wurtzite AlN. -

Gas-Liquid-Solid Condensation of Gravitationally Confined Polystyrene Particles Near a Charged Wall at Intermediate Screening Lengths
Int. J. Microgravity Sci. Appl. 32 (2) 2015, 320207 DOI :10.15011/ijmsa.32.320207 IIIII The Kikuchi-Kossel Experiment - Colloidal Crystals under Microgravity IIIII (Original Paper) Gas-Liquid-Solid Condensation of Gravitationally Confined Polystyrene Particles near a Charged Wall at Intermediate Screening Lengths Masamichi ISHIKAWA 1, Shun ITO 2, Saki MIYOSHI 3 and Junko HABASAKI 4 Abstract Charged polystyrene particles, which were initially sedimented uniformly on a glass wall, spontaneously transformed into two- dimensional gas, liquid, and solid phases in aqueous solvents. Their relative stabilities could be determined by a common phase diagram consisting of gas-liquid and liquid-solid coexistence lines under the conditions of a large particle radius (a >> κ1) and intermediate screening length (a=1.5 μm and 10 nm<κ1<300 nm), where a is the particle radius and κ is the Debye-Hückel parameter. The gas-liquid miscibility gap which had a critical point was very stable over a wide range of salt concentrations. The radial distribution functions of colloidal particles in the liquid phase showed remarkable oscillatory damping and both short-range (~particle diameter σ) and long-range (>10σ) decay lengths were found. The latter was attributed to the charge density wave (CDW) due to alternating layers of oppositely charged colloids and counterions along the radial coordinate. Because the charged glass wall had a similar surface charge density and sign as the particles, the repulsive levitation at the wall against gravity assisted the thermal rearrangement during phase separation. Further evaluation is promising for investigating the complicated physics of particle-wall interactions. Keyword(s): Polystyrene, Charged colloid, Colloidal phase transition, Sedimentation. -

1000 J Mole Μa ( ) = 4,0001.09 TJ Mole Μb
3.012 PS 6 THERMODYANMICS SOLUTIONS 3.012 Issued: 11.28.05 Fall 2005 Due: THERMODYNAMICS 1. Building a binary phase diagram. Given below are data for a binary system of two materials A and B. The two components form three phases: an ideal liquid solution, and two solid phases α and β that exhibit regular solution behavior. Use the given thermodynamic data to answer the questions below. LIQUID PHASE: ALPHA PHASE: L J " J µo = "500 " 4.8T µo = #6000 ( A ) mole ( A ) mole L J " J µo = "1,000 "10T µo = #1,000 ( B ) mole ( B ) mole ! ! (T is temperature in K) Ω = 8,368 J/mole ! ! BETA PHASE: " J µo = #4,000 #1.09T ( A ) mole " J µo = #13,552 # 2.09T ( B ) mole ! (T is temperature in K) ! Ω = 7,000 J/mole a. Plot the free energy of L, alpha, and beta phases (overlaid on one plot) vs. XB at the following temperatures: 2000K, 1500K, 1000K, 925K, and 800K. For each plot, denote common tangents and drop vertical lines below the plot to a ‘composition bar’, as illustrated in the example plot below. For each graph, mark the stable phases as a function of XB in the composition bar below the graph, as illustrated in the example. 3.012 PS 6 1 of 18 12/11/05 EXAMPLE: 3.012 PS 6 2 of 18 12/11/05 3.012 PS 6 3 of 18 12/11/05 b. Using the free energy curves prepared in part (a), construct a qualitatively correct phase diagram for this system in the temperature range 800K – 2000K. -

Chapter 10 Gibbs Free Energy Composition and Phase Diagrams of Binary Systems 10.2 Gibbs Free Energy and Thermodynamic Activity
Chapter 10 Gibbs Free Energy Composition and Phase Diagrams of Binary Systems 10.2 Gibbs Free Energy and Thermodynamic Activity The Gibbs free energy of mixing of the components A and B to form a mole of solution: If it is ideal, i.e., if ai=Xi, then the molar Gibbs free energy of mixing, Figure 10.1 The molar Gibbs free energies of Figure 10.2 The activities of component B mixing in binary systems exhibiting ideal behavior obtained from lines I, II, and III in Fig. 10.1. (I), positive deviation from ideal behavior (II), and negative deviation from ideal behavior (III). 10.3 The Gibbs Free Energy of Formation of Regular Solutions If curves II and III for regular solutions, then deviation of from , is ∆ ∆ For curve II, < , , and thus is a positive quantity ( and Ω are positive quantities). ∆ ∆ ∆ Figure 10.3 The effect of the magnitude of a on the integral molar heats and integral molar Gibbs free energies of formation of a binary regular solution. 10.3 The Gibbs Free Energy of Formation of Regular Solutions The equilibrium coexistence of two separate solutions at the temperature T and pressure P requires that and Subtracting from both sides of Eq. (i) gives 0 or Similarly Figure 10.4 (a) The molar Gibbs free energies of mixing of binary components which form a complete range of solutions. (b) The molar Gibbs free energies of mixing of binary components in a system which exhibits a miscibility gap. 10.4 Criteria for Phase Stability in Regular Solutions For a regular solution, and 3 3 At XA=XB=0.5, the third derivative, / XB =0, 2 2 and thus the second derivative, / XB =0 at XA=0.5 ∆ When α=2 ⇒ the critical value of ∆αabove which phase separation occurs. -
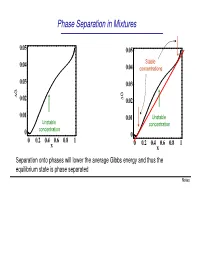
Phase Separation in Mixtures
Phase Separation in Mixtures 0.05 0.05 0.04 Stable 0.04 concentrations 0.03 0.03 G G ∆ 0.02 ∆ 0.02 0.01 0.01 Unstable Unstable concentration concentration 0 0 0 0.2 0.4 0.6 0.8 1 0 0.2 0.4 0.6 0.8 1 x x Separation onto phases will lower the average Gibbs energy and thus the equilibrium state is phase separated Notes Phase Separation versus Temperature Note that at higher temperatures the region of concentrations where phase separation takes place shrinks and G eventually disappears T increases ∆G = ∆U −T∆S This is because the term -T∆S becomes large at high temperatures. You can say that entropy “wins” over the potential energy cost at high temperatures Microscopically, the kinetic energy becomes x much larger than potential at high T, and the 1 x2 molecules randomly “run around” without noticing potential energy and thus intermix Oil and water mix at high temperature Notes Liquid-Gas Phase Separation in a Mixture A binary mixture can exist in liquid or gas phases. The liquid and gas phases have different Gibbs potentials as a function of mole fraction of one of the components of the mixture At high temperatures the Ggas <Gliquid because the entropy of gas is greater than that of liquid Tb2 T At lower temperatures, the two Gibbs b1 potentials intersect and separation onto gas and liquid takes place. Notes Boiling of a binary mixture Bubble point curve Dew point If we have a liquid at pint A that contains F A gas curve a molar fraction x1 of component 1 and T start heating it: E D - First the liquid heats to a temperature T’ C at constant x A and arrives to point B.