Targeting the FMS-Like Tyrosine Kinase 3 in Acute Myeloid Leukemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

How I Treat Myelofibrosis
From www.bloodjournal.org by guest on October 7, 2014. For personal use only. Prepublished online September 16, 2014; doi:10.1182/blood-2014-07-575373 How I treat myelofibrosis Francisco Cervantes Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Advance online articles have been peer reviewed and accepted for publication but have not yet appeared in the paper journal (edited, typeset versions may be posted when available prior to final publication). Advance online articles are citable and establish publication priority; they are indexed by PubMed from initial publication. Citations to Advance online articles must include digital object identifier (DOIs) and date of initial publication. Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved. From www.bloodjournal.org by guest on October 7, 2014. For personal use only. Blood First Edition Paper, prepublished online September 16, 2014; DOI 10.1182/blood-2014-07-575373 How I treat myelofibrosis By Francisco Cervantes, MD, PhD, Hematology Department, Hospital Clínic, IDIBAPS, University of Barcelona, Barcelona, Spain Correspondence: Francisco Cervantes, MD, Hematology Department, Hospital Clínic, Villarroel 170, 08036 Barcelona, Spain. Phone: +34 932275428. -

Thrombopoietin Supports the Continuous Growth of Cytokine-Dependent Human Leukemia Cell Lines HG Drexler, M Zaborski and H Quentmeier
Leukemia (1997) 11, 541–551 1997 Stockton Press All rights reserved 0887-6924/97 $12.00 Thrombopoietin supports the continuous growth of cytokine-dependent human leukemia cell lines HG Drexler, M Zaborski and H Quentmeier DSMZ-German Collection of Microorganisms and Cell Cultures, Department of Human and Animal Cell Cultures, Mascheroder Weg 1 B, D-38124 Braunschweig, Germany Hematopoiesis is a complex process of regulated cellular pro- nate membrane receptor. This binding triggers a series of intra- liferation and differentiation from the primitive stem cells to the cellular mediators involved in the growth factor’s signaling final fully differentiated cell. The long and extensive search for a factor specifically regulating megakaryocytopoiesis led to the pathways. Recently, a novel hematopoietic growth factor, cloning of a hormone, here called thrombopoietin (TPO), that termed thrombopoietin (TPO), was cloned and shown to be a specifically promotes proliferation and differentiation of the megakaryocytic lineage-associated growth and differentiation megakaryocytic lineage. The availability of recombinant TPO factor. Binding of TPO to its receptor, c-MPL, mediates plei- and its imminent clinical use has made a more detailed under- otropic effects on megakaryocyte development in vitro and in standing of its effects on hematopoietic cells more urgent. Nor- vivo. TPO is clearly the primary regulator of this cell lineage mal megakaryocyto- and thrombopoiesis occurs predomi- nantly in the bone marrow, a difficult organ to study in situ, acting at all levels of megakaryocytopoiesis and thrombopo- particularly in humans, due to the low numbers of megakary- iesis (reviewed in Ref. 1). ocytic progenitors and the consequent difficult isolation as The availability of TPO will be of considerable clinical pure populations. -

Therapeutic Class Overview Colony Stimulating Factors
Therapeutic Class Overview Colony Stimulating Factors Therapeutic Class Overview/Summary: This review will focus on the granulocyte colony stimulating factors (G-CSFs) and granulocyte- macrophage colony stimulating factors (GM-CSFs).1-5 Colony-stimulating factors (CSFs) fall under the naturally occurring glycoprotein cytokines, one of the main groups of immunomodulators.6 In general, these proteins are vital to the proliferation and differentiation of hematopoietic progenitor cells.6-8 The G- CSFs commercially available in the United States include pegfilgrastim (Neulasta®), filgrastim (Neupogen®), filgrastim-sndz (Zarxio®), and tbo-filgrastim (Granix®). While filgrastim-sndz and tbo- filgrastim are the same recombinant human G-CSF as filgrastim, only filgrastim-sndz is considered a biosimilar drug as it was approved through the biosimilar pathway. At the time tbo-filgrastim was approved, a regulatory pathway for biosimilar drugs had not yet been established in the United States and tbo-filgrastim was filed under its own Biologic License Application.9 Only one GM-CSF is currently available, sargramostim (Leukine). These agents are Food and Drug Administration (FDA)-approved for a variety of conditions relating to neutropenia or for the collection of hematopoietic progenitor cells for collection by leukapheresis.1-5 Due to the pathway taken, tbo-filgrastim does not share all of the same indications as filgrastim and these two products are not interchangeable. It is important to note that although filgrastim-sndz is a biosimilar product, and it was approved with all the same indications as filgrastim at the time, filgrastim has since received FDA-approval for an additional indication that filgrastim-sndz does not have, to increase survival in patients with acute exposure to myelosuppressive doses of radiation.1-3A complete list of indications for each agent can be found in Table 1. -

Side Effects of Molecular-Targeted Therapies in Solid Cancers : a New Challenge in Cancer Therapy Management
Side effects of molecular-targeted therapies in solid cancers : a new challenge in cancer therapy management Ahmad Awada, MD, PhD Medical Oncology Clinic Institut Jules Bordet Université Libre de Bruxelles (U.L.B.) Brussels, Belgium PLAN OF THE LECTURE 1. Concept 2. Achievements on the management of side effects 3. Remaining challenges 4. New challenges with the development of molecular-targeted therapies 5. Conclusions Reducing the cancer- related problems and the side effects of the SUPPORTIVE CARE = medicine administered to treat the disease SIDE EFFECTS OF CANCER THERAPY: ACHIEVEMENTS Side effect Preventive & Therapeutic intervention • Febrile neutropenia • G-CSF, Anti-infectives • Anemia • Epoetine •Mucositis •Laser therapy, Palifermin • Nausea & Vomiting • 5-HT3 and neurokin-1-receptor antagonists •Thromboembolic •LMW Heparin events • Cardiomyopathy • Liposomal formulations, Dexrazonane (anthracyclines) MANAGEMENT OF SIDE EFFECTS : REMAINING CHALLENGES • Alopecia • Thrombocytopenia ( ! Promising Thrombopoietin- mimetics are under investigation) • Asthenia MOLECULAR TARGETS AND THERAPIES (1) Drug Class Mechanism of action Main tumor indication Gefitinib* Small molecule TK inhibitor of EGFR NSCLC (Iressa) Erlotinib* Small molecule TK inhibitor of EGFR NSCLC (Tarceva) Cetuximab* Monoclonal Antibody Blocks EGFR Colorectal, Head & (Erbitux) Neck, NSCLC Monoclonal Panitumumab* Antibody Blocks EGFR Colorectal (Vectibix) * Investigational in BC TK : tyrosine kinase; EGFR : epidermal growth factor receptor MOLECULAR TARGETS AND THERAPIES -

DOPTELET (Avatrombopag) RATIONALE for INCLUSION IN
DOPTELET (avatrombopag) RATIONALE FOR INCLUSION IN PA PROGRAM Background Doptelet is a thrombopoietin (TPO) receptor agonist used to increase platelet counts. Doptelet (avatrombopag) is an orally bioavailable, small molecule TPO receptor agonist that stimulates proliferation and differentiation of megakaryocytes from bone marrow progenitor cells resulting in an increased production of platelets. Doptelet does not compete with TPO for binding to the TPO receptor and has an additive effect with TPO on platelet production (1). Regulatory Status FDA approved indication: Doptelet is a thrombopoietin receptor agonist indicated for the treatment of: (1) 1. Thrombocytopenia in adult patients with chronic liver disease who are scheduled to undergo a procedure. 2. Thrombocytopenia in adult patients with chronic immune thrombocytopenia who have had an insufficient response to a previous treatment. Doptelet should not be administered to patients with chronic liver disease in an attempt to normalize platelet counts (1). Doptelet is a thrombopoietin (TPO) receptor agonist and TPO receptor agonists have been associated with thrombotic and thromboembolic complications in patients with chronic liver disease. A Doppler ultrasound is a noninvasive test that can be used to estimate the blood flow through blood vessels by bouncing high-frequency sound waves (ultrasound) off circulating red blood cells. A Doppler ultrasound may help determine if Doptelet therapy is appropriate for a patient (1-2). The safety and effectiveness of Doptelet in pediatric patients have not been established (1). Summary Doptelet is a thrombopoietin (TPO) receptor agonist used to increase platelet counts. Doptelet (avatrombopag) is an orally bioavailable, small molecule TPO receptor agonist that stimulates proliferation and differentiation of megakaryocytes from bone marrow progenitor cells resulting in an increased production of platelets. -

Insights Into the Cellular Mechanisms of Erythropoietin-Thrombopoietin Synergy
Papayannopoulou et al.: Epo and Tpo Synergy Experimental Hematology 24:660-669 (19961 661 @ 1996 International Society for Experimental Hematology Rapid Communication ulation with fluorescence microscopy. Purified subsets were grown in plasma clot and methylcellulose clonal cultures and in suspension cultures using the combinations of cytokines Insights into the cellular mechanisms cadaveric bone marrow cells obtained from Northwest described in the text. Single cells from the different subsets Center, Puget Sound Blood Bank (Seattle, WA), were were. also deposited (by FACS) on 96-well plates containing of erythropoietin-thrombopoietin synergy washed, and incubated overnight in IMDM with 10% medmm and cytokines. Clonal growth from single-cell wells calf serum on tissue culture plates to remove adherent were double-labeled with antiglycophorin A-PE and anti Thalia Papayannopoulou, Martha Brice, Denise Farrer, Kenneth Kaushansky From the nonadherent cells, CD34+ cells were isolated CD41- FITC between days 10 and 19. direct immunoadherence on anti-CD34 monoclonal anti University of Washington, Department of Medicine, Seattle, WA (mAb)-coated plates, as previously described [15]. Purity Immunocytochemistry Offprint requests to: Thalia Papayannopoulou, MD, DrSci, University of Washington, isolated CD34+ cells ranged from 80 to 96% by this For immunocytochemistry, either plasma clot or cytospin cell Division of Hematology, Box 357710, Seattle, WA 98195-7710 od. Peripheral blood CD34 + cells from granulocyte preparations were used. These were fixed at days 6-7 and (Received 24 January 1996; revised 14 February 1996; accepted 16 February 1996) ulating factor (G-CSF)-mobilized normal donors 12-13 with pH 6.5 Histochoice (Amresco, Solon, OH) and provided by Dr. -

FLT3 Inhibitors in Acute Myeloid Leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2*
Wu et al. Journal of Hematology & Oncology (2018) 11:133 https://doi.org/10.1186/s13045-018-0675-4 REVIEW Open Access FLT3 inhibitors in acute myeloid leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2* Abstract FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents. Keywords: FMS-like tyrosine kinase 3 inhibitors, Acute myeloid leukemia, Midostaurin, FLT3 Introduction RAS, MEK, and PI3K/AKT pathways [10], and ultim- Acute myeloid leukemia (AML) remains a highly resist- ately causes suppression of apoptosis and differentiation ant disease to conventional chemotherapy, with a me- of leukemic cells, including dysregulation of leukemic dian survival of only 4 months for relapsed and/or cell proliferation [11]. refractory disease [1]. Molecular profiling by PCR and Multiple FLT3 inhibitors are in clinical trials for treat- next-generation sequencing has revealed a variety of re- ing patients with FLT3/ITD-mutated AML. In this re- current gene mutations [2–4]. New agents are rapidly view, we summarized the preclinical and clinical studies emerging as targeted therapy for high-risk AML [5, 6]. on new FLT3 inhibitors, including sorafenib, lestaurtinib, In 1996, FMS-like tyrosine kinase 3/internal tandem du- sunitinib, tandutinib, quizartinib, midostaurin, gilteriti- plication (FLT3/ITD) was first recognized as a frequently nib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG mutated gene in AML [7]. -

Comparison Between Filgrastim and Lenograstim Plus Chemotherapy For
Bone Marrow Transplantation (2010) 45, 277–281 & 2010 Macmillan Publishers Limited All rights reserved 0268-3369/10 $32.00 www.nature.com/bmt ORIGINAL ARTICLE Comparison between filgrastim and lenograstim plus chemotherapy for mobilization of PBPCs R Ria, T Gasparre, G Mangialardi, A Bruno, G Iodice, A Vacca and F Dammacco Department of Biomedical Sciences and Human Oncology, Section of Internal Medicine and Clinical Oncology, University of Bari Medical School, Bari, Italy Recombinant human (rHu) G-CSF has been widely used during transplantation.4 However, the use of G-CSF after to treat neutropenia and mobilize PBPCs for their autologous PBPC transplantation has been queried, as its autologous and allogeneic transplantation. It shortens further reduction in time to a safe neutrophil count5,6 does neutropenia and thus reduces the frequency of neutropenic not always imply fewer significant clinical events, such as fever. We compared the efficiency of glycosylated rHu infections, length of hospitalization, extrahematological and non-glycosylated Hu G-CSF in mobilizing hemato- toxicities or mortality.7,8 Even so, the ASCO guidelines still poietic progenitor cells (HPCs). In total, 86 patients were recommend the use of growth factors after autologous consecutively enrolled for mobilization with CY plus either PBPC transplantation.9 glycosylated or non-glycosylated G-CSF, and under- G-CSF induces the proliferation and differentiation of went leukapheresis. The HPC content of each collection, myeloid precursor cells, and also provides a functional toxicity, days of leukapheresis needed to reach the activity that influences chemotaxis, respiratory burst and minimum HPC target and days to recover WBC (X500 Ag expression of neutrophils. -

Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling While Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells
Published OnlineFirst February 14, 2013; DOI: 10.1158/1535-7163.MCT-12-0305 Molecular Cancer Cancer Therapeutics Insights Therapeutics Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling while Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells Ruwanthi N. Gunawardane, Ronald R. Nepomuceno, Allison M. Rooks, Jeremy P. Hunt, Jill M. Ricono, Barbara Belli, and Robert C. Armstrong Abstract Fms-like tyrosine kinase 3 (FLT3) is implicated in the pathogenesis of acute myeloid leukemia (AML). FLT3-activating internal tandem duplication (ITD) mutations are found in approximately 30% of patients with AML and are associated with poor outcome in this patient population. Quizartinib (AC220) has previously been shown to be a potent and selective FLT3 inhibitor. In the current study, we expand on previous observations by showing that quizartinib potently inhibits the phosphorylation of FLT3 and downstream signaling molecules independent of FLT3 genotype, yet induces loss of viability only in cells expressing constitutively activated FLT3. We further show that transient exposure to quizartinib, whether in vitro or in vivo, leads to prolonged inhibition of FLT3 signaling, induction of apoptosis, and drastic reductions in tumor volume and pharmacodynamic endpoints. In vitro experiments suggest that these prolonged effects are mediated by slow binding kinetics that provide for durable inhibition of the kinase following drug removal/clearance. Together these data suggest quizartinib, with its unique combination of selectivity and potent/sustained inhibition of FLT3, may provide a safe and effective treatment against FLT3-driven leukemia. Mol Cancer Ther; 12(4); 1–10. Ó2013 AACR. Introduction downstream signaling cascades including the Ras/ Fms-like tyrosine kinase 3 (FLT3) is a member of the MAPK, PI3K/Akt, and STAT5 pathways (1, 9–13). -
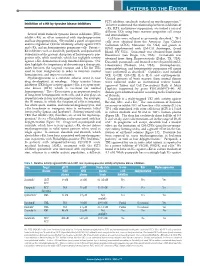
Letters to the Editor
LETTERS TO THE EDITOR FLT3 inhibitor, sorafenib, induced no myelosuppression.5,6 Inhibition of c-Kit by tyrosine kinase inhibitors To better understand the relationship between inhibition of c-Kit, FLT3, and marrow suppression, we studied a series of different TKIs using bone marrow progenitor cell assays Several small molecule tyrosine kinase inhibitors (TKIs) and immunoblots. inhibit c-Kit, an effect associated with myelosuppression Cell lines were cultured as previously described.2 TF-1 and hair depigmentation. We studied a panel of approved cells were obtained from the American Type Culture and investigational TKIs for inhibitory activity against FLT3 Collection (ATCC; Manassas, VA, USA) and grown in and c-Kit, and on hematopoietic progenitor cells. Potent c- RPMI supplemented with GM-CSF (Invitrogen, Grand Kit inhibitors such as dasatinib, pazopanib, and quizartinib Island, NY, USA). Quizartinib was obtained from Ambit demonstrated the greatest disruption of hematopoietic pro- Biosciences (San Diego, CA, USA). Crenolanib was genitor cells, while sorafenib, which has negligible activity obtained from Arog Pharmaceuticals (Dallas, TX, USA). against c-Kit, demonstrated only minimal disruption. Our Dasatinib, pazopanib, and imatinib were obtained from LC data highlight the importance of determining a therapeutic Laboratories (Woburn, MA, USA). Electrophoresis, index between the targeted receptor and c-Kit for TKIs immunoblotting, and hematopoietic progenitor cell assays used to treat malignancies in order to maintain normal were performed as described.2 Cytokines used included hematopoiesis and improve outcomes. SCF, G-CSF, GM-CSF, IL-3, IL-6, and erythropoietin. Myelosuppression is a common adverse event in new Unused portions of bone marrow from normal donors drug development in oncology. -

Cytokine Signaling in Tumor Progression
Immune Netw. 2017 Aug;17(4):214-227 https://doi.org/10.4110/in.2017.17.4.214 pISSN 1598-2629·eISSN 2092-6685 Review Article Cytokine Signaling in Tumor Progression Myungmi Lee, Inmoo Rhee* Department of Bioscience and Biotechnology, Sejong University, Seoul 05006, Korea Received: Apr 13, 2017 ABSTRACT Revised: Jun 22, 2017 Accepted: Jun 25, 2017 Cytokines are molecules that play critical roles in the regulation of a wide range of normal *Correspondence to functions leading to cellular proliferation, differentiation and survival, as well as in Inmoo Rhee specialized cellular functions enabling host resistance to pathogens. Cytokines released Department of Bioscience and Biotechnology, in response to infection, inflammation or immunity can also inhibit cancer development Sejong University, 209 Neungdong-ro, and progression. The predominant intracellular signaling pathway triggered by cytokines Gwangjin-gu, Seoul 05006, Korea. is the JAK-signal transducer and activator of transcription (STAT) pathway. Knockout mice Tel: +82-2-6935-2432 E-mail: [email protected] and clinical human studies have provided evidence that JAK-STAT proteins regulate the immune system, and maintain immune tolerance and tumor surveillance. Moreover, aberrant Copyright © 2017. The Korean Association of activation of the JAK-STAT pathways plays an undeniable pathogenic role in several types Immunologists of human cancers. Thus, in combination, these observations indicate that the JAK-STAT This is an Open Access article distributed under the terms of the Creative Commons proteins are promising targets for cancer therapy in humans. The data supporting this view Attribution Non-Commercial License (https:// are reviewed herein. creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial Keywords: Cytokine; JAK-STAT; Cancer; Kinase inhibitor use, distribution, and reproduction in any medium, provided the original work is properly cited. -

Human G-CSF ELISA Kit (ARG80143)
Product datasheet [email protected] ARG80143 Package: 96 wells Human G-CSF ELISA Kit Store at: 4°C Component Cat. No. Component Name Package Temp ARG80143-001 Antibody-coated 8 X 12 strips 4°C. Unused strips microplate should be sealed tightly in the air-tight pouch. ARG80143-002 Standard 3 X 2 ng/vial 4°C (Lyophilized) ARG80143-003 Standard diluent 20 ml 4°C buffer ARG80143-004 Antibody conjugate 400 µl 4°C concentrate ARG80143-005 Antibody diluent 16 ml 4°C buffer ARG80143-006 HRP-Streptavidin 400 µl 4°C (Protect from concentrate light) ARG80143-007 HRP-Streptavidin 16 ml 4°C diluent buffer ARG80143-008 20X Wash buffer 50 ml 4°C ARG80143-009 TMB substrate 12ml 4°C (Protect from light) ARG80143-010 STOP solution 12ml 4°C ARG80143-011 Plate sealer 4 strips Room temperature Summary Product Description ARG80143 Human G-CSF ELISA Kit is an Enzyme Immunoassay kit for the quantification of Human G-CSF in Serum, Plasma, Cell culture supernatants. Tested Reactivity Hu Tested Application ELISA Specificity No significant cross-reactivity or interference with Human CT-1,IL-11,OSM,CNTF,HGF,M-CSF;mouse IL-6;rat IL-6,CNTF Target Name G-CSF Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm Sensitivity 15 pg/ml Sample Type Serum, Plasma, Cell culture supernatants Standard Range 31.25 - 2000 pg/ml www.arigobio.com 1/3 Sample Volume 100 µl Precision CV: Alternate Names Granulocyte colony-stimulating factor; Lenograstim; C17orf33; GCSF; G-CSF; Filgrastim; Pluripoietin; CSF3OS Application Instructions Assay Time 4 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C.