Genome-Wide Association Study Reveals Two New Risk Loci for Bipolar Disorder
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
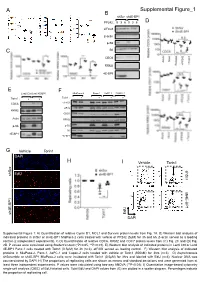
H Supplemental Figure 1 I D E B a C
A Supplemental Figure_1 B shScr sh4E-BP1 PP242 : 0 3 6 0 3 6 D eIF4G1 β-actin p-S6 C S6 CDC6 RRM2 4E-BP1 E F Lenti Ctrl Lenti 4E-BP1 MiaPaca-2 Panc-1 AsPC-1 Capan-2 Torin1 : Torin1 : + + + + + + CDC6 eIF4G1 eIF3a RRM2 CDC6 Actin RRM2 p-S6 p-S6 S6 4E-BP1 4E-BP1 G Vehicle Torin1 DAPI H I Vehicle Torin1 62.20% 45.30% shScr sh4E-BP1 EdU shScr 59.58% 49.47% EdU sh4E-BP1 DAPI Supplemental Figure 1: A) Quantification of relative Cyclin D1, MCL1 and Survivin protein levels from Fig. 1A. B) Western blot analysis of indicated proteins in shScr or sh4E-BP1 MiaPaca-2 cells treated with vehicle or PP242 (5µM) for 3h and 6h. β-actin served as a loading control (2 independent experiments). C-D) Quantification of relative CDC6, RRM2 and CDC7 protein levels from (C) Fig. 2C and (D) Fig. 2D. P values were calculated using Student’s t-test (*P<0.05, **P<0.01). E) Western blot analysis of indicated proteins in Lenti Ctrl or Lenti 4E-BP1 Panc-1 cells treated with Torin1 (0.5µM) for 3h (n=3). eIF4GI served as loading control. F) Western blot analysis of indicated proteins in MiaPaca-2, Panc-1, AsPC-1 and Capan-2 cells treated with vehicle or Torin1 (500nM) for 3hrs (n=3). G) Asynchronous shScramble or sh4E-BP1 MiaPaca-2 cells were incubated with Torin1 (0.5μM) for 3hrs and labeled with EdU (n=3). Nuclear DNA was counterstained by DAPI. H) The proportions of replicating cells are shown as means and standard deviations and were generated from at least three independent experiments. -

MTHFR) and Methionine Synthase Reductase (MTRR
ANTICANCER RESEARCH 28 : 2807-2812 (2008) Methylenetetrahy drofolate Reductase (MTHFR) and Methionine Synthase Reductase (MTRR) Gene Polymorphisms as Risk Factors for Hepatocellular Carcinoma in a Korean Population SUN YOUNG KWAK 1,2 , UN KYUNG KIM 3, HYO JIN CHO 2, HEE KEUN LEE 3, HYE JIN KIM 2, NAM KEUN KIM 2 and SEONG GYU HWANG 1,2 1Department of Internal Medicine, 2Institute for Clinical Research, College of Medicine, Pochon CHA University, Seongnam; 3Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, South Korea Abstract. Hepatocellular carcinoma (HCC) is the third were increased risk factors for the disease. The MTHFR most frequent cause of cancer death in South Korea, but 1298A>C and the MTRR 66A>G genotypes were associated genetic susceptibility factors of HCC have not been examined with an increased risk of HCC in th is Korean population. extensively. Methylenetetrahydrofolate reductase (MTHFR) Further studies involving larger and varied populations and methionine synthase reductase (MTRR) play an essential could provide a potential tool for cancer risk assessment in role in both DNA synthesis and methylation and patients who are at risk of developing HCC. polymorphisms in the MTHFR gene, 677C>T, 1298A>C and the MTRR gene, 66A>G, are associated with several types of Hepatocellular carcinoma (HCC) is one of the most common malignancy. In this study, the allelic frequencies and malignant tumors with the third highest mortality rate among genotype distribution of three polymorphisms in the MTHFR malignancies in South Korea. According to a statistical report and MTRR genes from 96 hepatocellular carcinoma (HCC) in 2006, it occurred in 33.8/10,000 men and 11.2/10,000 patients and 201 controls were examined to assess the women in South Korea in 2005 (1). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
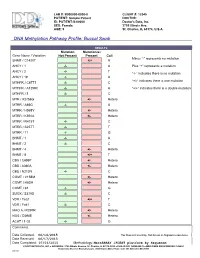
DNA Methylation Pathway Profile; Buccal Swab
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-00000 Doctor's Data, Inc. SEX: Female 3755 Illinois Ave. AGE: 5 St. Charles, IL 60174, U.S.A. !DNA Methylation Pathway Profile; Buccal Swab RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call Minus “-“ represents no mutation SHMT / C1420T +/+ A AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A “+/-“ indicates there is one mutation MTHFR / C677T -/- C MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G -/- A MTRR / H595Y +/- Hetero MTRR / K350A +/- Hetero MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 -/- G BHMT / 1 -/- A BHMT / 2 -/- C BHMT / 4 +/- Hetero BHMT / 8 +/+ T CBS / C699T +/- Hetero CBS / A360A +/- Hetero CBS / N212N -/- C COMT / V158M +/- Hetero COMT / H62H +/- Hetero COMT / 61 -/- G SUOX / S370S -/- C VDR / Taq1 +/+ T VDR / Fok1 -/- C MAO A / R297R +/- Hetero NOS / D298E +/- Hetero ACAT / 1-02 -/- G Comments: Date Collected: 06/14/2015 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 06/17/2015 Date Completed: 07/03/2015 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

Chicken Linkage Disequilibrium Is Much More Complex Over Much Longer Distance Than Previously Appreciated
Look Over the Horizon - Chicken Linkage Disequilibrium is Much More Complex Over Much Longer Distance than Previously Appreciated Ehud Lipkin ( [email protected] ) Hebrew University of Jerusalem Janet E. Fulton Hy-Line (United States) Jacqueline Smith University of Edinburgh David W. Burt University of Edinburgh Morris Soller Hebrew University of Jerusalem Research Article Keywords: Chicken, long-range linkage disequilibrium, QTL, F6, LD blocks Posted Date: June 17th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-598396/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract Background Appreciable Linkage Disequilibrium (LD) is commonly found between pairs of loci close to one another, decreasing rapidly with distance between the loci. This provides the basis studies to map Quantitative Trait Loci Regions (QTLRs), where it is custom to assume that the closest sites to a signicant markers are the prime candidate to be the causative mutation. Nevertheless, Long-Range LD (LRLD) can also be found among well-separated sites. LD blocks are runs of genomic sites all having appreciable LD with one another. High LD and LRLD are often separated by genomic sites with which they have practically no LD. Thus, not only can LD be found among distant loci, but also its pattern may be complex, comprised of fragmented blocks. Here, chicken LRLD and LD blocks, and their relationship with previously described Marek’s Disease (MD) QTLRs, were studied in an F6 population from a full-sib advanced intercross line, and in eight commercial pure layer lines. -

The Transition from Primary Colorectal Cancer to Isolated Peritoneal Malignancy
medRxiv preprint doi: https://doi.org/10.1101/2020.02.24.20027318; this version posted February 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . The transition from primary colorectal cancer to isolated peritoneal malignancy is associated with a hypermutant, hypermethylated state Sally Hallam1, Joanne Stockton1, Claire Bryer1, Celina Whalley1, Valerie Pestinger1, Haney Youssef1, Andrew D Beggs1 1 = Surgical Research Laboratory, Institute of Cancer & Genomic Science, University of Birmingham, B15 2TT. Correspondence to: Andrew Beggs, [email protected] KEYWORDS: Colorectal cancer, peritoneal metastasis ABBREVIATIONS: Colorectal cancer (CRC), Colorectal peritoneal metastasis (CPM), Cytoreductive surgery and heated intraperitoneal chemotherapy (CRS & HIPEC), Disease free survival (DFS), Differentially methylated regions (DMR), Overall survival (OS), TableFormalin fixed paraffin embedded (FFPE), Hepatocellular carcinoma (HCC) ARTICLE CATEGORY: Research article NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2020.02.24.20027318; this version posted February 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . NOVELTY AND IMPACT: Colorectal peritoneal metastasis (CPM) are associated with limited and variable survival despite patient selection using known prognostic factors and optimal currently available treatments. -

Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from in Vitro Model Systems
International Journal of Molecular Sciences Review Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems Ritu Nayak, Idan Rosh, Irina Kustanovich and Shani Stern * Sagol Department of Neurobiology, University of Haifa, Haifa 3498838, Israel; [email protected] (R.N.); [email protected] (I.R.); [email protected] (I.K.) * Correspondence: [email protected] Abstract: Bipolar disorder (BD) and schizophrenia are psychiatric disorders that manifest unusual mental, behavioral, and emotional patterns leading to suffering and disability. These disorders span heterogeneous conditions with variable heredity and elusive pathophysiology. Mood stabilizers such as lithium and valproic acid (VPA) have been shown to be effective in BD and, to some extent in schizophrenia. This review highlights the efficacy of lithium and VPA treatment in several randomized, controlled human trials conducted in patients suffering from BD and schizophrenia. Furthermore, we also address the importance of using induced pluripotent stem cells (iPSCs) as a disease model for mirroring the disease’s phenotypes. In BD, iPSC-derived neurons enabled finding an endophenotype of hyperexcitability with increased hyperpolarizations. Some of the disease phenotypes were significantly alleviated by lithium treatment. VPA studies have also reported rescuing the Wnt/β-catenin pathway and reducing activity. Another significant contribution of iPSC models can be attributed to studying the molecular etiologies of schizophrenia such as abnormal differentiation of patient-derived neural stem cells, decreased neuronal connectivity and neurite Citation: Nayak, R.; Rosh, I.; number, impaired synaptic function, and altered gene expression patterns. Overall, despite significant Kustanovich, I.; Stern, S. Mood advances using these novel models, much more work remains to fully understand the mechanisms Stabilizers in Psychiatric Disorders by which these disorders affect the patients’ brains. -

Signature Redacted Thesis Supervisor Certified By
Single-Cell Transcriptomics of the Mouse Thalamic Reticular Nucleus by Taibo Li S.B., Massachusetts Institute of Technology (2015) Submitted to the Department of Electrical Engineering and Computer Science in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2017 @ Massachusetts Institute of Technology 2017. All rights reserved. A uthor ... ..................... Department of Electrical Engineering and Computer Science May 25, 2017 Certified by. 3ignature redacted Guoping Feng Poitras Professor of Neuroscience, MIT Signature redacted Thesis Supervisor Certified by... Kasper Lage Assistant Professor, Harvard Medical School Thesis Supervisor Accepted by . Signature redacted Christopher Terman Chairman, Masters of Engineering Thesis Committee MASSACHUSETTS INSTITUTE 0) OF TECHNOLOGY w AUG 14 2017 LIBRARIES 2 Single-Cell Transcriptomics of the Mouse Thalamic Reticular Nucleus by Taibo Li Submitted to the Department of Electrical Engineering and Computer Science on May 25, 2017, in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science Abstract The thalamic reticular nucleus (TRN) is strategically located at the interface between the cortex and the thalamus, and plays a key role in regulating thalamo-cortical in- teractions. Current understanding of TRN neurobiology has been limited due to the lack of a comprehensive survey of TRN heterogeneity. In this thesis, I developed an integrative computational framework to analyze the single-nucleus RNA sequencing data of mouse TRN in a data-driven manner. By combining transcriptomic, genetic, and functional proteomic data, I discovered novel insights into the molecular mecha- nisms through which TRN regulates sensory gating, and suggested targeted follow-up experiments to validate these findings. -

DNA Methylation Pathway Profile; Blood Spot
LAB #: B000000-0000-0 CLIENT #: 12345 PATIENT: Sample Patient DOCTOR: ID: PATIENT-S-000000000 Doctor's Data, Inc. SEX: Male 3755 Illinois Ave AGE: 45 St. Charles, IL 60174 USA !DNA Methylation Pathway Profile; Blood Spot RESULTS Mutation Mutation(s) Gene Name / Variation Not Present Present Call SHMT / C1420T +/- Hetero Minus “-“ represents no mutation AHCY / 1 -/- A Plus “+” represents a mutation AHCY / 2 -/- T “-/-“ indicates there is no mutation AHCY / 19 -/- A MTHFR / C677T -/- C “+/-“ indicates there is one mutation MTHFR / A1298C -/- A “+/+” indicates there is a double mutation MTHFR / 3 -/- C MTR / A2756G +/- Hetero MTRR / A66G +/- Hetero MTRR / H595Y -/- C MTRR / K350A -/- A MTRR / R415T -/- C MTRR / S257T -/- T MTRR / 11 +/- Hetero BHMT / 1 -/- A BHMT / 2 +/- Hetero BHMT / 4 +/+ C BHMT / 8 +/+ T CBS / C699T -/- C CBS / A360A -/- C CBS / N212N -/- C COMT / V158M +/+ A COMT / H62H +/+ T COMT / 61 -/- G SUOX / S370S -/- CG VDR / Taq1 -/- C VDR / Fok1 -/- C MAO A / R297R -/- G NOS / D298E -/- G ACAT / 1-02 +/- Hetero Comments: Date Collected: 11/08/2012 *For Research Use Only. Not for use in diagnostic procedures. Date Received: 11/13/2012 Date Completed: 12/10/2012 Methodology: MassARRAY iPLEXT platform by Sequenom ©DOCTOR’S DATA, INC. !!! ADDRESS: 3755 Illinois Avenue, St. Charles, IL 60174-2420 !!! CLIA ID NO: 14D0646470 !!! MEDICARE PROVIDER NO: 148453 Analyzed by Bioserve Biotechnologies, 9000 Virginia Manor Road, Suite 207, Beltsville, MD 20705 0001867 Methionine Metabolism Transmethylation & Transsulfuration Diagram Lab number: B000000-0000-0 DNA Methylatn BldSpt Page: 1 Patient: Sample Patient Client: 12345 Introduction Single nucleotide polymorphisms (SNPs) are DNA sequence variations, which may occur frequently in the population (at least one percent of the population.) They are different from disease mutations, which are very rare. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Analysis of Seven Maternal Polymorphisms of Genes Involved In
July 2006 ⅐ Vol. 8 ⅐ No. 7 article Analysis of seven maternal polymorphisms of genes involved in homocysteine/folate metabolism and risk of Down syndrome offspring Iris Scala, MD1, Barbara Granese, PhD1, Maria Sellitto, MD1, Serena Salome`, MD1, Annalidia Sammartino, MD2, Antonio Pepe, PhD1, Pierpaolo Mastroiacovo, MD3, Gianfranco Sebastio, MD1, and Generoso Andria, MD1 PURPOSE: We present a case-control study of seven polymorphisms of six genes involved in homocysteine/folate pathway as risk factors for Down syndrome. Gene-gene/allele-allele interactions, haplotype analysis and the association with age at conception were also evaluated. METHODS: We investigated 94 Down syndrome-mothers and 264 control-women from Campania, Italy. RESULTS: Increased risk of Down syndrome was associated with the methylenetetrahydrofolate reductase (MTHFR) 1298C allele (OR 1.46; 95% CI 1.02–2.10), the MTHFR 1298CC genotype (OR 2.29; 95% CI 1.06–4.96), the reduced-folate-carrier1 (RFC1) 80G allele (1.48; 95% CI 1.05–2.10) and the RFC1 80 GG genotype (OR 2.05; 95% CI 1.03–4.07). Significant associations were found between maternal age at conception Ն34 years and either the MTHFR 1298C or the RFC 180G alleles. Positive interactions were found for the following genotype-pairs: MTHFR 677TT and 1298CC/CA, 1298CC/CA and RFC1 80 GG/GA, RFC1 80 GG and methylenetetrahydrofolate-dehydrogenase 1958 AA. The 677–1298 T-C haplotype at the MTHFR locus was also a risk factor for Down syndrome (P ϭ 0.0022). The methionine-synthase-reductase A66G, the methionine-synthase A2756G and the cystathionine-beta-synthase 844ins68 polymorphisms were not associated with increased risk of Down syndrome.