Oligomerization of ß-Dystroglycan in Rabbit Diaphragm and Brain As Revealed by Chemical Crosslinking
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Role of Z-Disc Proteins in Myopathy and Cardiomyopathy
International Journal of Molecular Sciences Review The Role of Z-disc Proteins in Myopathy and Cardiomyopathy Kirsty Wadmore 1,†, Amar J. Azad 1,† and Katja Gehmlich 1,2,* 1 Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK; [email protected] (K.W.); [email protected] (A.J.A.) 2 Division of Cardiovascular Medicine, Radcliffe Department of Medicine and British Heart Foundation Centre of Research Excellence Oxford, University of Oxford, Oxford OX3 9DU, UK * Correspondence: [email protected]; Tel.: +44-121-414-8259 † These authors contributed equally. Abstract: The Z-disc acts as a protein-rich structure to tether thin filament in the contractile units, the sarcomeres, of striated muscle cells. Proteins found in the Z-disc are integral for maintaining the architecture of the sarcomere. They also enable it to function as a (bio-mechanical) signalling hub. Numerous proteins interact in the Z-disc to facilitate force transduction and intracellular signalling in both cardiac and skeletal muscle. This review will focus on six key Z-disc proteins: α-actinin 2, filamin C, myopalladin, myotilin, telethonin and Z-disc alternatively spliced PDZ-motif (ZASP), which have all been linked to myopathies and cardiomyopathies. We will summarise pathogenic variants identified in the six genes coding for these proteins and look at their involvement in myopathy and cardiomyopathy. Listing the Minor Allele Frequency (MAF) of these variants in the Genome Aggregation Database (GnomAD) version 3.1 will help to critically re-evaluate pathogenicity based on variant frequency in normal population cohorts. -

Influence of Nebulin on the Assembly Mechanics Of
INFLUENCE OF NEBULIN ON THE ASSEMBLY MECHANICS OF DESMIN An Undergraduate Research Scholars Thesis by MARC ADRIAN CARAGEA Submitted to Honors and Undergraduate Research Texas A&M University In partial fulfillment of the requirements for the designation as UNDERGRADUATE RESEARCH SCHOLAR Approved by Research Adviser: Gloria M. Conover, Ph.D. May 2014 Major: Biomedical Sciences TABLE OF CONTENTS Page ABSTRACT ............................................................................................................................... 1 ACKNOWLEDGMENTS ........................................................................................................... 2 NOMENCLATURE .................................................................................................................... 3 CHAPTER I INTRODUCTION ............................................................................................... 4 II MATERIALS AND METHODS ......................................................................... 8 Affinity purification of WT and mutant desmin proteins from bacteria ................ 8 Affinity purification of recombinant Nebulin M160 – 164 ................................... 9 Assembly protocol for desmin precursors into filaments in vitro ...................... 11 Sample preparation for atomic force microscopy ............................................... 11 Single desmin filament length acquisition and analysis ..................................... 13 Determination of Young’s modulus for desmin filament networks ................... -

Profiling of the Muscle-Specific Dystroglycan Interactome Reveals the Role of Hippo Signaling in Muscular Dystrophy and Age-Dependent Muscle Atrophy Andriy S
Yatsenko et al. BMC Medicine (2020) 18:8 https://doi.org/10.1186/s12916-019-1478-3 RESEARCH ARTICLE Open Access Profiling of the muscle-specific dystroglycan interactome reveals the role of Hippo signaling in muscular dystrophy and age-dependent muscle atrophy Andriy S. Yatsenko1†, Mariya M. Kucherenko2,3,4†, Yuanbin Xie2,5†, Dina Aweida6, Henning Urlaub7,8, Renate J. Scheibe1, Shenhav Cohen6 and Halyna R. Shcherbata1,2* Abstract Background: Dystroglycanopathies are a group of inherited disorders characterized by vast clinical and genetic heterogeneity and caused by abnormal functioning of the ECM receptor dystroglycan (Dg). Remarkably, among many cases of diagnosed dystroglycanopathies, only a small fraction can be linked directly to mutations in Dg or its regulatory enzymes, implying the involvement of other, not-yet-characterized, Dg-regulating factors. To advance disease diagnostics and develop new treatment strategies, new approaches to find dystroglycanopathy-related factors should be considered. The Dg complex is highly evolutionarily conserved; therefore, model genetic organisms provide excellent systems to address this challenge. In particular, Drosophila is amenable to experiments not feasible in any other system, allowing original insights about the functional interactors of the Dg complex. Methods: To identify new players contributing to dystroglycanopathies, we used Drosophila as a genetic muscular dystrophy model. Using mass spectrometry, we searched for muscle-specific Dg interactors. Next, in silico analyses allowed us to determine their association with diseases and pathological conditions in humans. Using immunohistochemical, biochemical, and genetic interaction approaches followed by the detailed analysis of the muscle tissue architecture, we verified Dg interaction with some of the discovered factors. -

Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics
International Journal of Molecular Sciences Review Troponin Variants in Congenital Myopathies: How They Affect Skeletal Muscle Mechanics Martijn van de Locht , Tamara C. Borsboom, Josine M. Winter and Coen A. C. Ottenheijm * Department of Physiology, Amsterdam Cardiovascular Sciences, Amsterdam UMC, Location VUmc, 1081 HZ Amsterdam, The Netherlands; [email protected] (M.v.d.L.); [email protected] (T.C.B.); [email protected] (J.M.W.) * Correspondence: [email protected]; Tel.: +31-(0)-20-444-8123 Abstract: The troponin complex is a key regulator of muscle contraction. Multiple variants in skeletal troponin encoding genes result in congenital myopathies. TNNC2 has been implicated in a novel congenital myopathy, TNNI2 and TNNT3 in distal arthrogryposis (DA), and TNNT1 and TNNT3 in nemaline myopathy (NEM). Variants in skeletal troponin encoding genes compromise sarcomere function, e.g., by altering the Ca2+ sensitivity of force or by inducing atrophy. Several potential therapeutic strategies are available to counter the effects of variants, such as troponin activators, introduction of wild-type protein through AAV gene therapy, and myosin modulation to improve muscle contraction. The mechanisms underlying the pathophysiological effects of the variants in skeletal troponin encoding genes are incompletely understood. Furthermore, limited knowledge is available on the structure of skeletal troponin. This review focusses on the physiology of slow and fast skeletal troponin and the pathophysiology of reported variants in skeletal troponin encoding genes. A better understanding of the pathophysiological effects of these variants, together with enhanced knowledge regarding the structure of slow and fast skeletal troponin, will direct the development of Citation: van de Locht, M.; treatment strategies. -

Distal Myopathies a Review: Highlights on Distal Myopathies with Rimmed Vacuoles
Review Article Distal myopathies a review: Highlights on distal myopathies with rimmed vacuoles May Christine V. Malicdan, Ikuya Nonaka Department of Neuromuscular Research, National Institutes of Neurosciences, National Center of Neurology and Psychiatry, Tokyo, Japan Distal myopathies are a group of heterogeneous disorders Since the discovery of the gene loci for a number classiÞ ed into one broad category due to the presentation of distal myopathies, several diseases previously of weakness involving the distal skeletal muscles. The categorized as different disorders have now proven to recent years have witnessed increasing efforts to identify be the same or allelic disorders (e.g. distal myopathy the causative genes for distal myopathies. The identiÞ cation with rimmed vacuoles and hereditary inclusion body of few causative genes made the broad classiÞ cation of myopathy, Miyoshi myopathy and limb-girdle muscular these diseases under “distal myopathies” disputable and dystrophy type 2B (LGMD 2B). added some enigma to why distal muscles are preferentially This review will focus on the most commonly affected. Nevertheless, with the clariÞ cation of the molecular known and distinct distal myopathies, using a simple basis of speciÞ c conditions, additional clues have been classification: distal myopathies with known molecular uncovered to understand the mechanism of each condition. defects [Table 1] and distal myopathies with unknown This review will give a synopsis of the common distal causative genes [Table 2]. The identification of the myopathies, presenting salient facts regarding the clinical, genes involved in distal myopathies has broadened pathological, and molecular aspects of each disease. Distal this classification into sub-categories as to the location myopathy with rimmed vacuoles, or Nonaka myopathy, will of encoded proteins: sarcomere (titin, myosin); plasma be discussed in more detail. -

Effect of Nebulin Variants on Nebulin-Actin Interaction
Pro gradu –thesis EFFECT OF NEBULIN VARIANTS ON NEBULIN-ACTIN INTERACTION Svetlana Sofieva November 2019 University of Helsinki Genetics Folkhälsan research center Specialization in Human Genetics Tiedekunta – Fakultet – Faculty Laitos – Institution– Department Faculty of biological and environmental sciences Department of Biosciences Tekijä – Författare – Author Svetlana Sofieva Työn nimi – Arbetets titel – Title Effect of nebulin variants on nebulin-actin interaction Oppiaine – Läroämne – Subject Human genetics Työn laji – Arbetets art – Level Aika – Datum – Month and year Sivumäärä – Sidoantal – Number of pages Master’s thesis 11/2019 61 pages + 15 pages in Appendix Tiivistelmä – Referat – Abstract Nemaline myopathy (NM) is a rare congenital disorder, the most common of congenital myopathies. It affects primarily the skeletal muscles and it is recognised by nemaline bodies in muscle tissue samples and muscle weakness. Mutation of eleven genes are known to lead to NM and the most frequent disease-causing variants are either recessive NEB variants or dominant ACTA1 variants. Variants in NEB are thought to be well tolerated and only 7% of them are hypothesized to be pathogenic. Over 200 pathogenic NEB- variants have been identified in Helsinki and the majority occurred in patients as a combination of two different variants. The missense variants were speculated to have a modifying effect on pathogenicity by affecting nebulin-actin or nebulin-tropomyosin interactions. Nebulin is a gigantic protein coded by NEB and is one of the largest proteins in vertebrates. It is located in the thin filament of the skeletal muscle sarcomere. Enclosed by terminal regions, nebulin has an extensive repetitive modular region that covers over 90% of the protein. -

Diagnosis and Cell-Based Therapy for Duchenne Muscular Dystrophy in Humans, Mice, and Zebrafish
J Hum Genet (2006) 51:397–406 DOI 10.1007/s10038-006-0374-9 MINIREVIEW Louis M. Kunkel Æ Estanislao Bachrach Richard R. Bennett Æ Jeffrey Guyon Æ Leta Steffen Diagnosis and cell-based therapy for Duchenne muscular dystrophy in humans, mice, and zebrafish Received: 3 January 2006 / Accepted: 4 January 2006 / Published online: 1 April 2006 Ó The Japan Society of Human Genetics and Springer-Verlag 2006 Abstract The muscular dystrophies are a heterogeneous mutants carries a stop codon mutation in dystrophin, group of genetically caused muscle degenerative disor- and we have recently identified another carrying a ders. The Kunkel laboratory has had a longstanding mutation in titin. We are currently positionally cloning research program into the pathogenesis and treatment of the disease-causative mutation in the remaining 12 mu- these diseases. Starting with our identification of dys- tant strains. We hope that one of these new mutant trophin as the defective protein in Duchenne muscular strains of fish will have a mutation in a gene not previ- dystrophy (DMD), we have continued our work on ously implicated in human muscular dystrophy. This normal dystrophin function and how it is altered in gene would become a candidate gene to be analyzed in muscular dystrophy. Our work has led to the identifi- patients which do not carry a mutation in any of the cation of the defective genes in three forms of limb girdle known dystrophy-associated genes. By studying both muscular dystrophy (LGMD) and a better understand- disease pathology and investigating potential therapies, ing of how muscle degenerates in many of the different we hope to make a positive difference in the lives of dystrophies. -

Regulation of Titin-Based Cardiac Stiffness by Unfolded Domain Oxidation (Undox)
Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx) Christine M. Loeschera,1, Martin Breitkreuzb,1, Yong Lia, Alexander Nickelc, Andreas Ungera, Alexander Dietld, Andreas Schmidte, Belal A. Mohamedf, Sebastian Kötterg, Joachim P. Schmitth, Marcus Krügere,i, Martina Krügerg, Karl Toischerf, Christoph Maackc, Lars I. Leichertj, Nazha Hamdanib, and Wolfgang A. Linkea,2 aInstitute of Physiology II, University of Munster, 48149 Munster, Germany; bInstitute of Physiology, Ruhr University Bochum, 44801 Bochum, Germany; cComprehensive Heart Failure Center Wuerzburg, University Clinic Wuerzburg, 97078 Wuerzburg, Germany; dDepartment of Internal Medicine II, University Hospital Regensburg, 93053 Regensburg, Germany; eInstitute for Genetics, University of Cologne, 50931 Cologne, Germany; fDepartment of Cardiology and Pneumology, University Medicine Goettingen, 37075 Goettingen, Germany; gDepartment of Cardiovascular Physiology, Heinrich Heine University, 40225 Düsseldorf, Germany; hDepartment of Pharmacology and Clinical Pharmacology, Heinrich Heine University, 40225 Düsseldorf, Germany; iCenter for Molecular Medicine and Excellence Cluster "Cellular Stress Responses in Aging-Associated Diseases" (CECAD), University of Cologne, 50931 Cologne, Germany; and jInstitute for Biochemistry and Pathobiochemistry, Ruhr University Bochum, 44801 Bochum, Germany Edited by Jonathan Seidman, Harvard University, Boston, MA, and approved August 12, 2020 (received for review March 14, 2020) The relationship between oxidative stress and -

Neuronal Dystroglycan Is Necessary for Formation and Maintenance of Functional CCK-Positive Basket Cell Terminals on Pyramidal Cells
10296 • The Journal of Neuroscience, October 5, 2016 • 36(40):10296–10313 Cellular/Molecular Neuronal Dystroglycan Is Necessary for Formation and Maintenance of Functional CCK-Positive Basket Cell Terminals on Pyramidal Cells Simon Fru¨h,1,5 Jennifer Romanos,1,5 Patrizia Panzanelli,2 XDaniela Bu¨rgisser,3 XShiva K. Tyagarajan,1,5 X Kevin P. Campbell,4 XMirko Santello,1,5 and XJean-Marc Fritschy1,5 1Institute of Pharmacology and Toxicology, University of Zurich, 8057 Zurich, Switzerland, 2Department of Neuroscience Rita Levi Montalcini, University of Turin, 10124 Turin, Italy, 3ETH Zurich, 8092 Zurich, Switzerland, 4Howard Hughes Medical Institute, Department of Molecular Physiology and Biophysics, Department of Neurology, University of Iowa Roy J. and Lucille A. Carver College of Medicine, Iowa City, Iowa 52242, and 5Neuroscience Center Zurich, University of Zurich and ETH Zurich, 8057 Zurich, Switzerland Distinct types of GABAergic interneurons target different subcellular domains of pyramidal cells, thereby shaping pyramidal cell activity patterns. Whether the presynaptic heterogeneity of GABAergic innervation is mirrored by specific postsynaptic factors is largely unex- plored. Here we show that dystroglycan, a protein responsible for the majority of congenital muscular dystrophies when dysfunctional, has a function at postsynaptic sites restricted to a subset of GABAergic interneurons. Conditional deletion of Dag1, encoding dystrogly- can, in pyramidal cells caused loss of CCK-positive basket cell terminals in hippocampus and neocortex. PV-positive basket cell terminals were unaffected in mutant mice, demonstrating interneuron subtype-specific function of dystroglycan. Loss of dystroglycan in pyrami- dal cells had little influence on clustering of other GABAergic postsynaptic proteins and of glutamatergic synaptic proteins. -

Postmortem Changes in the Myofibrillar and Other Cytoskeletal Proteins in Muscle
BIOCHEMISTRY - IMPACT ON MEAT TENDERNESS Postmortem Changes in the Myofibrillar and Other C'oskeletal Proteins in Muscle RICHARD M. ROBSON*, ELISABETH HUFF-LONERGAN', FREDERICK C. PARRISH, JR., CHIUNG-YING HO, MARVIN H. STROMER, TED W. HUIATT, ROBERT M. BELLIN and SUZANNE W. SERNETT introduction filaments (titin), and integral Z-line region (a-actinin, Cap Z), as well as proteins of the intermediate filaments (desmin, The cytoskeleton of "typical" vertebrate cells contains paranemin, and synemin), Z-line periphery (filamin) and three protein filament systems, namely the -7-nm diameter costameres underlying the cell membrane (filamin, actin-containing microfilaments, the -1 0-nm diameter in- dystrophin, talin, and vinculin) are listed along with an esti- termediate filaments (IFs), and the -23-nm diameter tubu- mate of their abundance, approximate molecular weights, lin-containing microtubules (Robson, 1989, 1995; Robson and number of subunits per molecule. Because the myofibrils et al., 1991 ).The contractile myofibrils, which are by far the are the overwhelming components of the skeletal muscle cell major components of developed skeletal muscle cells and cytoskeleton, the approximate percentages of the cytoskel- are responsible for most of the desirable qualities of muscle eton listed for the myofibrillar proteins (e.g., myosin, actin, foods (Robson et al., 1981,1984, 1991 1, can be considered tropomyosin, a-actinin, etc.) also would represent their ap- the highly expanded corollary of the microfilament system proximate percentages of total myofibrillar protein. of non-muscle cells. The myofibrils, IFs, cell membrane skel- eton (complex protein-lattice subjacent to the sarcolemma), Some Important Characteristics, Possible and attachment sites connecting these elements will be con- Roles, and Postmortem Changes of Key sidered as comprising the muscle cell cytoskeleton in this Cytoskeletal Proteins review. -

Α-Actinin/Titin Interaction: a Dynamic and Mechanically Stable Cluster of Bonds in the Muscle Z-Disk
α-Actinin/titin interaction: A dynamic and mechanically stable cluster of bonds in the muscle Z-disk Marco Grisona, Ulrich Merkela, Julius Kostanb, Kristina Djinovic-Carugob,c, and Matthias Riefa,d,1 aPhysik Department E22, Technische Universität München, 85748 Garching, Germany; bDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, A-1030 Vienna, Austria; cDepartment of Biochemistry, Faculty of Chemistry and Chemical Technology, University of Ljubljana, SI-1000 Ljubljana, Slovenia; and dMunich Center for Integrated Protein Science, 81377 Munich, Germany Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved December 16, 2016 (received for review August 2, 2016) Stable anchoring of titin within the muscle Z-disk is essential for In humans, the isoforms of titin exhibit four to seven Z-repeats preserving muscle integrity during passive stretching. One of the (15, 16, 20). The structure of the EF3-4 hands complex with titin main candidates for anchoring titin in the Z-disk is the actin cross- Z-repeat 7 shows the bound Z-repeat in an α-helical confor- linker α-actinin. The calmodulin-like domain of α-actinin binds to mation (21). In solution assays, binding affinities of various the Z-repeats of titin. However, the mechanical and kinetic prop- Z-repeats to EF3-4 were determined to lie in the micromolar erties of this important interaction are still unknown. Here, we use range (22). Micromolar affinity points only to a moderately a dual-beam optical tweezers assay to study the mechanics of this stable interaction, the kinetics of which are unknown. -
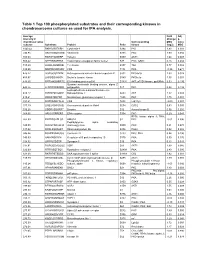
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin