A High-Performance Liquid Chromatography Assay Method for the Determination of Lidocaine in Human Serum
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
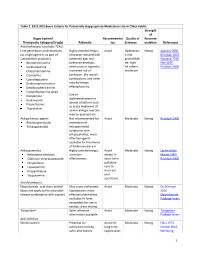
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

NORPACE- Disopyramide Phosphate Capsule, Gelatin Coated
NORPACE- disopyramide phosphate capsule, gelatin coated Carilion Materials Management ---------- Norpace disopyramide phosphate capsules Norpace CR disopyramide phosphate extended- release capsules ® ® DESCRIPTION Norpace (disopyramide phosphate) is an antiarrhythmic drug available for oral administration in immediate-release and controlled-release capsules containing 100 mg or 150 mg of disopyramide base, present as the phosphate. The base content of the phosphate salt is 77.6%. The structural formula of Norpace is: Norpace is freely soluble in water, and the free base (pKa 10.4) has an aqueous solubility of 1 mg/ml. The chloroform:water partition coefficient of the base is 3.1 at pH 7.2. Norpace is a racemic mixture of and isomers. This drug is not chemically related to other antiarrhythmic drugs. d-l- Norpace CR (controlled-release) capsules are designed to afford a gradual and consistent release of disopyramide. Thus, for maintenance therapy, Norpace CR provides the benefit of less-frequent dosing (every 12 hours) as compared with the every-6-hour dosage schedule of immediate-release Norpace capsules. Inactive ingredients of Norpace include corn starch, edible ink, FD&C Red No. 3, FD&C Yellow No. 6, gelatin, lactose, talc, and titanium dioxide; the 150-mg capsule also contains FD&C Blue No. 1. Inactive ingredients of Norpace CR include corn starch, D&C Yellow No. 10, edible ink, ethylcellulose, FD&C Blue No. 1, gelatin, shellac, sucrose, talc, and titanium dioxide; the 150-mg capsule also contains FD&C Red No. 3 and FD&C Yellow No. 6. CLINICAL PHARMACOLOGY Mechanisms of Action Norpace (disopyramide phosphate) is a Type 1 antiarrhythmic drug (i.e., similar to procainamide and quinidine). -

Disopyramide for Obstructive Hypertrophic Cardiomyopathy Symptoms and Gradient Resistant to First-Line Medical Therapy Approxima
Disopyramide for Obstructive Hypertrophic Cardiomyopathy Symptoms and Gradient Resistant to First‐Line Medical Therapy Approximately 2/3 of patients with hypertrophic cardiomyopathy (HCM) have left ventricular outflow tract (LVOT) obstruction either at rest or after physiologic provocation. Besides left ventricular hypertrophy (LVH), such patients have either resting, or provocable LVOT gradient > 30mmHg which contributes to their symptoms of exercise intolerance, dyspnea, angina or syncope. Moreover, resting gradient is associated with decreased survival. The most common cause of LVOT obstruction is due to systolic anterior motion of the mitral valve (SAM) and mitral‐septal contact. The underlying cause of SAM is an altered internal geometry of the LV, leading to an overlap between the inflow and outflow portions of the LV. Besides septal hypertrophy, this overlap is caused by anterior displacement of the mitral apparatus (papillary muscles and mitral leaflets) and mitral slack. Drag, the pushing force of flow is the dominant hydrodynamic force that causes SAM; flow gets behind the mitral leaflets and sweeps them into the septum. LVOT obstruction is associated with increased systolic LV work, decreased diastolic aortic perfusion pressure, supply‐demand ischemia, load‐related impairment in diastolic relaxation and a mid‐systolic drop in instantaneous LV ejection flow velocities and flow 1 A unique feature of obstructive HCM is the provocable gradient. Obstruction worsens after physiologic stimuli that reduce preload and afterload and increase contractility such as Valsalva maneuver, standing, after eating and particularly after exercise. Unfortunately, the more widely used cardiac medications, such as ACE‐inhibitors, ARBs, vasodilators, and nitrates are deleterious in exactly this way and increase gradient because of their vasodilatory properties. -

Neemmc Guidelines for Tablet Crushing and Administration Via Enteral Feeding Tubes
NEEMMC GUIDELINES FOR TABLET CRUSHING AND ADMINISTRATION VIA ENTERAL FEEDING TUBES KEY TO DRUG ADMINISTRATION GUIDELINES Please follow the guidelines in order, as shown in the chart (i.e. number 1 is the first choice of which form to administer the drug in). A Tablet will disperse in 1-2 minutes. B Tablet will disperse in greater than 2 minutes. C Liquid preparation available. D Dilute reconstituted injection with 30-60ml of water before administering. E Oral solution/suspension can be prepared by local pharmacy or Non Sterile Preparative Services (PSU at Colchester General Hospital). Note: It is an unlicensed use to crush tablets, open capsules and make extemporaneous suspensions. However, using tablets within these guidelines is covered by the Trust for legal/vicarious liability. For more information on the administration via different tubes, please contact Medicines Information on ext. 2161. Drug Key code Information ACETAZOLAMIDE 1. A* * Diamox SR capsules can be opened and 2. D the contents flushed down the enteral tube 3. E ACICLOVIR 1. C * Dispersible tablets available 2. A* ALFACALCIDOL C* * Drops available. Sensitive to light ALFUZOSIN A Beware of sudden hypotensive effect if giving crushed tablets. Monitor BP and ensure patient is lying down prior to administering the dose. Do not crush slow release preparations. ALLOPURINOL 1. B 2. E ALVERINE Content of capsules is very bitter, and might numb the tongue and throat. AMILORIDE 1. B 2. C* * Liquid available. AMINOPHYLLINE Convert to theophylline liquid equivalent. Do not crush SR preparations. AMIODARONE B AMITRIPTYLINE 1. C 2. B AMLODIPINE A A Tablet will disperse in 1-2 minutes. -

DISOPYRAMIDE PHOSPHATE CAPSULES, Usp31273129rx Only
DISOPYRAMIDE PHOSPHATE- disopyramide phosphate capsule Carilion Materials Management ---------- DISOPYRAMIDE PHOSPHATE CAPSULES, USP 3127 3129 Rx only DESCRIPTION Disopyramide Phosphate is an antiarrhythmic drug available for oral administration in capsules containing 100 mg or 150 mg of disopyramide base, present as the phosphate. The base content of the phosphate salt is 77.6%. The structural formula of Disopyramide Phosphate is: C21H29N3O•H3PO4 M.W. 437.47 α-[2-(diisopropylamino) ethyl]-α-phenyl-2-pyridineacetamide phosphate Disopyramide Phosphate is freely soluble in water, and the free base (pKa 10.4) has an aqueous solubility of 1 mg/mL. The chloroform:water partition coefficient of the base is 3.1 at pH 7.2. Disopyramide Phosphate is a racemic mixture of d- and l-isomers. This drug is not chemically related to other antiarrhythmic drugs. Inactive Ingredients:Capsules: Lactose Monohydrate, Magnesium Stearate and Sodium Starch Glycolate. Capsule Print and Shell Constituents: Black Iron Oxide, D&C Red #28, D&C Red #33, D&C Yellow #10, D&C Yellow #10 Aluminum Lake, FD&C Blue #1, FD&C Blue #1 Aluminum Lake, FD&C Blue #2 Aluminum Lake, FD&C Red #40 Aluminum Lake, Gelatin, Propylene Glycol, Shellac, Sodium Lauryl Sulfate, Sorbitan Monolaurate and Titanium Dioxide. CLINICAL PHARMACOLOGY Mechanisms of Action Disopyramide Phosphate is a Type 1 antiarrhythmic drug (i.e., similar to procainamide and quinidine). In animal studies, Disopyramide Phosphate decreases the rate of diastolic depolarization (phase 4) in cells with augmented automaticity, decreases the upstroke velocity (phase 0) and increases the action potential duration of normal cardiac cells, decreases the disparity in refractoriness between infarcted and adjacent normally perfused myocardium, and has no effect on alpha- or beta-adrenergic receptors. -

DOMASYS Standard
NEW ZEALAND DATA SHEET 1 PRODUCT NAME Rythmodan 100mg capsules Rythmodan 150mg# capsules #Not marketed 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Rythmodan 100mg and 150mg# capsules contain 100mg and 150mg of disopyramide respectively For the full list of excipients, see section 6.1. 3 PHARMACEUTICAL FORM 100 mg capsules: green & beige, marked RY RL 150 mg# capsules: white, marked RY 150 in black. 4 CLINICAL PARTICULARS 4.1 THERAPEUTIC INDICATIONS Disopyramide is indicated for the treatment of symptomatic and/or life-threatening arrhythmias. Specific indications are as follows: Disopyramide is indicated in the following symptomatic arrhythmias: • atrial or ventricular extrasystoles • treatment of atrial arrhythmias • atrial fibrillation or flutter: preparation for cardioversion; substitution for cardioversion, when the latter is contraindicated or unfeasible; prevention of relapses following restoration of sinus rhythm rythmodan-ccdsv6-dsv7-31may21 Page 1 • prevention of attacks of paroxysmal tachycardia, Bouveret type, including Wolff-Parkinson- White syndrome • prevention of relapses of paroxysmal ventricular tachycardia 4.2 DOSE AND METHOD OF ADMINISTRATION The daily dose of RYTHMODAN capsules must be administered as no less than 3 equal divided doses. Adults with Normal Hepatic and Renal Function: Prevention or Curative Treatment of Symptomatic Arrhythmias Initial treatment: 400 to 600 mg per day Maintenance treatment: reached by progressively reducing the daily dose by 100 mg per day to reach a final dose of 300 to 400 mg per day on average. Substitution Treatment for Cardioversion When cardioversion is contraindicated or otherwise not feasible, the initial capsule dosage can be increased to 800 mg on the first day in 4 divided doses. Preparation for cardioversion, after stopping digitalis 100 mg capsule on the previous evening 3 x 100 mg just before cardioversion in three doses at hourly intervals 100 mg capsule on the evening of cardioversion followed by the usual maintenance treatment from the next day onwards. -

Antiarrhythmic Pharmacotherapy
Antiarrhythmic Pharmacopoeia Powerful drugs, split into 4 major classes, according to the predominant receptor they effect. Some fit into several classes at once, like sotolol. Some don’t fit at all, owing to the intrinsic weirdness of their actions and the archaic quaintness of this system of classification, introduced in 1970 by EM Vaughan Williams. CLASS 1 drugs: Fast Sodium Channel Blockers, “membrane stabilizers” These are split into 3 subclasses, organized by binding speed (but, curiously, not in any order). The classes are 1a, 1b and 1c. From fastest to slowest binding, the order is B, A, C. Why would you want different speeds? The speed of binding is also the speed of dissociation from the receptor. Many of these drugs only bind to an active receptor. Lets say the heart is going very fast, and the drug is slow to dissociate from the receptor: each time an action potential comes past the cell, more and more channels are going to get blocked. This results in a general slowing of the rate of action potential propagation, and so the QRS interval widens. CLASS 1 A: quinidine, procainamide, and disopyramide. Used very rararely.rely. Only Disopyramide is available for use in Australia, and the rest have fallenfallen out of favour in 2007. “moderateAll Class 1a blockade” drugs have some potassium channel blockade capacity;- Block With fasta slow sodium heart channels,rate, they thuswill become reduce ratearrhythmogenic, of phase 0 upstroke because the potassium- Block blockade repolarising becomes potassium the dominant channels, feature. thus (prolong“reverse repolarisatiuse dependence”)on - Prolong QRS interval - Prolong QT interval Therefore, the length of the action potential is greatly slowed. -

Some Cardiovascular Problems with Disopyramide JOHN HAMER
Postgrad Med J: first published as 10.1136/pgmj.56.654.229 on 1 April 1980. Downloaded from Postgradiuate Medical Journal (April 1980) 56, 229-233 Some cardiovascular problems with disopyramide S. J. WARRINGTON JOHN HAMER M.A., M.R.C.P. M.D., Ph.D., F.R.C.P. Department of Clinical Pharmnacology, St Bartholomew's Hospital, London, E.C.I Summary review assessed the mortality of simple quinidine Five patients with apparent adverse cardiovascular conversion of atrial fibrillation as 3% (Thomson, effects of disopyramide are reviewed. Attention is 1956). drawn to the following problems. Four aspects of the cardiac effects of diso- (1) A vagolytic effect may produce a sinus tachy- pyramide noted by the authors recently seem worthy cardia with wide QRS complexes due to aberrant ofcomment: (1) adversecardiovasculareffects related conduction or intraventricular block superficially to the additional vagolytic effect of disopyramide, resembling a ventricular tachycardia, or may allow similar to those seen with quinidine; (2) adverse increased transmission of an atrial tachycardia or effects in sinus node disease; (3) a capacity to provoke atrial flutter to the ventricles by improving atrio- dangerous ventriculararrhythmias as is also seen with ventricular conduction. quinidine (Krikler and Curry, 1976); in addition, (2) Although the vagolytic effect is helpful in (4) the authors wish to stress the need for caution increasing sinus rate in patients with sinus node with intravenous injection to avoid side effects which disease, disopyramide may lead to bradycardia and may be related to immediate transient high plasma copyright. asystolic cardiac arrest, and should not be used concentrations before distribution of the drug can without a demand pacemaker. -
Ocular Side Effects of Disopyramide
Br J Ophthalmol: first published as 10.1136/bjo.68.12.890 on 1 December 1984. Downloaded from British Journal of Ophthalmology, 1984, 68, 890-891 Ocular side effects of disopyramide J. FRUCHT', I. FREIMANN,2 AND S. MERIN' From the Departments of ' Ophthalmology and 2Cardiology, Hadassah University Hospital, Kiryat Hadassah, Jerusalem, Israel SUMMARY A patient who suffered from severe decrease of accommodation andpupillarydilatation following the systemic use of disopyramide is described. The ocular side effects when this drug is used in large doses result from its anticholinergic action. Disopyramide (4 di-isopropylamino-2 phenyl-2-(2- tests were within the normal range. The patient had pyridyl) butyramide phosphate) is currently used as no ocular symptoms. an antiarrhythmic drug.' 2 Its pharmacological effect On her second day in hospital the attacks of ventri- on the heart is thought to be known.'-3 Disopyramide cular tachycardia no longer responded to the usual slows the depolarisation rate of the action potential, antiarrhythmic drugs and the cardiologist decided to depresses phase 4, and prolongs the duration of re- treat her with high doses of disopyramide. The polansation.' With the widespread use of the drug patient's weight was 49 kg. A dose of 200 mg of copyright. several adverse side effects have been described.4 disopyramide was injected intravenously and 150 mg Most were caused by its anticholinergic action and six times a day was given orally. On the next day the included dry mouth, urinary retention,5 and acute oral dose was raised to 300 mg six times a day. This angle closure glaucoma.6 Disopyramide has been re- dose finally prevented the attacks of ventricular ported47 to cause blurred vision, but no explanation tachycardia. -
Disopyramide (Norpace)
ALDERFER & TRAVIS CARDIOLOGY, PC 670 Lawn Ave., Suite 3A Sellersville, PA 18960 Tel (215)257-9500 FAX (215) 257-9500 DISOPYRAMIDE - ORAL OTHER NAMES: Norpace CR, Rythmodan USES: This medication is used to treat irregular heart rhythms (arrhythmias) and maintain a normal heart rate. HOW TO TAKE THIS MEDICATION: This is best taken with a full glass of water on an empty stomach one hour before or two hours after meals. If stomach upset occurs, it may be taken with food. Sustained-release tablets and capsules must be swallowed whole. Do not crush or chew them. This medication works best when there is a constant level of the drug in your body. To do this, take each dose as prescribed at evenly spaced intervals throughout the day and night. Try to take each dose at the same time each day. SIDE EFFECTS: Blurred vision, dry mouth, constipation, or difficulty urinating may occur as your body adjusts to the medication. If these symptoms persist or become bothersome, contact your doctor. Inform your doctor if you develop chest pain, rapid heartbeat, confusion, easy bruising or bleeding, rash, or breathing difficulties while taking this medication. This drug may cause dizziness and lightheadedness. To avoid this, rise from a seated or lying position slowly. Avoid alcohol PRECAUTIONS: Women who are pregnant or breast-feeding should inform their doctors. Before taking disopyramide, tell your doctor if you have myasthenia gravis; high blood pressure; glaucoma (or a family history of glaucoma); heart, liver, kidney, or prostate disease; difficulty urinating. Do not stop taking this medication without consulting your doctor. -
Emerging Medical Treatment for Hypertrophic Cardiomyopathy
Journal of Clinical Medicine Review Emerging Medical Treatment for Hypertrophic Cardiomyopathy Alessia Argirò 1,* , Mattia Zampieri 1, Martina Berteotti 1, Alberto Marchi 1, Luigi Tassetti 1 , Chiara Zocchi 1, Luisa Iannone 2, Beatrice Bacchi 2, Francesco Cappelli 1, Pierluigi Stefàno 2, Niccolò Marchionni 3 and Iacopo Olivotto 1 1 Cardiomyopathy Unit, Careggi University Hospital, 50134 Florence, Italy; [email protected] (M.Z.); [email protected] (M.B.); [email protected] (A.M.); [email protected] (L.T.); [email protected] (C.Z.); [email protected] (F.C.); [email protected] (I.O.) 2 Cardiac Surgery, Careggi University Hospital, 50134 Florence, Italy; [email protected] (L.I.); [email protected] (B.B.); pierluigi.stefano@unifi.it (P.S.) 3 Department of Experimental and Clinical Medicine, University of Florence, 50121 Florence, Italy; niccolo.marchionni@unifi.it * Correspondence: [email protected] Abstract: Hypertrophic cardiomyopathy (HCM) is a common myocardial disease characterized by otherwise unexplained left ventricular hypertrophy. The main cause of disabling symptoms in patients with HCM is left ventricular outflow tract (LVOT) obstruction. This phenomenon is multifactorial, determined both by anatomical and functional abnormalities: myocardial hyper- contractility is believed to represent one of its major determinants. The anatomical anomalies are targeted by surgical interventions, whereas attenuating hypercontractility is the objective of old and new drugs including the novel class of allosteric myosin inhibitors. This review summarizes the Citation: Argirò, A.; Zampieri, M.; current treatment modalities and discusses the emerging therapeutical opportunities focusing on the Berteotti, M.; Marchi, A.; Tassetti, L.; recently developed cardiac myosin ATPase inhibitors Mavacamten and CK-274. -
Pharmacokinetic Interactions Between Digoxin and Other Drugs
CORE Metadata, citation and similar papers at core.ac.uk Provided by82A Elsevier - Publisher Connector JACC Vol. 5, No.5 May 1985:82A-90A Pharmacokinetic Interactions Between Digoxin and Other Drugs FRANK I. MARCUS, MD, FACC Tucson, Arizona Drug interactions with digoxin are important because of particularly verapamil, have a similar effect. Potassium this agent's narrow therapeutic index. Amongthe drugs sparing diuretic drugs, such as spironolactone, can alter that can decrease digoxin bioavailability are cholestyr digoxin pharmacokinetics. amine, antacid gels, kaolin-peetate, certain antimicro Indomethacin may decrease renal excretion of dig bial drugs and cancer chemotherapeutic agents. In oxin in preterm infants. Finally, rifampin, an antibiotic selected patients, antibiotics may enhance digoxin bio used in the treatment of tuberculosis, may lower steady availability by eliminating intestinal flora that metabo state serum digoxin levels in patients with severe renal lize digoxin. disease. Physicians must maintain constant vigilance Antiarrhythmic drugs, such as quinidine and amio whenever medications are added to or withdrawn from darone, can markedly increase steady state serum dig a therapeutic regimen that includes digoxin. oxin levels. Certain calcium channel blocking drugs, (J Am Coil CardioI1985;5:82A-90A) Historical Perspective There is a high incidence of toxicity during the therapeutic use of this class of drugs. Interactions of cardiac glycosides Drug interactions with digoxin could only have been with other drugs contribute significantly to the high prev suspected but not verified before the availability of assays alence of digitalis toxicity. This review will focus on the sufficiently sensitive to measure digoxin in biological fluids pharmacokinetic interactions of digoxin.