Microarray-Based Comparative Genomic and Transcriptome Analysis of Borrelia Burgdorferi
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Longitudinal Expression Profiling of CD4+ and CD8+ Cells in Patients with Active to Quiescent Giant Cell Arteritis Elisabeth De Smit1*† , Samuel W
De Smit et al. BMC Medical Genomics (2018) 11:61 https://doi.org/10.1186/s12920-018-0376-4 RESEARCHARTICLE Open Access Longitudinal expression profiling of CD4+ and CD8+ cells in patients with active to quiescent giant cell arteritis Elisabeth De Smit1*† , Samuel W. Lukowski2†, Lisa Anderson3, Anne Senabouth2, Kaisar Dauyey2, Sharon Song3, Bruce Wyse3, Lawrie Wheeler3, Christine Y. Chen4, Khoa Cao4, Amy Wong Ten Yuen1, Neil Shuey5, Linda Clarke1, Isabel Lopez Sanchez1, Sandy S. C. Hung1, Alice Pébay1, David A. Mackey6, Matthew A. Brown3, Alex W. Hewitt1,7† and Joseph E. Powell2† Abstract Background: Giant cell arteritis (GCA) is the most common form of vasculitis affecting elderly people. It is one of the few true ophthalmic emergencies but symptoms and signs are variable thereby making it a challenging disease to diagnose. A temporal artery biopsy is the gold standard to confirm GCA, but there are currently no specific biochemical markers to aid diagnosis. We aimed to identify a less invasive method to confirm the diagnosis of GCA, as well as to ascertain clinically relevant predictive biomarkers by studying the transcriptome of purified peripheral CD4+ and CD8+ T lymphocytes in patients with GCA. Methods: We recruited 16 patients with histological evidence of GCA at the Royal Victorian Eye and Ear Hospital, Melbourne, Australia, and aimed to collect blood samples at six time points: acute phase, 2–3 weeks, 6–8 weeks, 3 months, 6 months and 12 months after clinical diagnosis. CD4+ and CD8+ T-cells were positively selected at each time point through magnetic-assisted cell sorting. RNA was extracted from all 195 collected samples for subsequent RNA sequencing. -
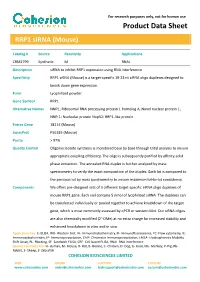
Product Data Sheet
For research purposes only, not for human use Product Data Sheet RRP1 siRNA (Mouse) Catalog # Source Reactivity Applications CRM2799 Synthetic M RNAi Description siRNA to inhibit RRP1 expression using RNA interference Specificity RRP1 siRNA (Mouse) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol RRP1 Alternative Names NNP1; Ribosomal RNA processing protein 1 homolog A; Novel nuclear protein 1; NNP-1; Nucleolar protein Nop52; RRP1-like protein Entrez Gene 18114 (Mouse) SwissProt P56183 (Mouse) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of mouse RRP1 gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Application key: E- ELISA, WB- Western blot, IH- Immunohistochemistry, IF- Immunofluorescence, -

Reading Targeted DNA Damage in the Active
Reading targeted DNA damage in the active demethylation pathway: role of accessory domains of eukaryotic AP endonucleases and thymine-DNA glycosylases Alexander Popov, Inga Grin, Antonina Dvornikova, Bakhyt Matkarimov, Regina Groisman, Murat Saparbaev, Dmitry Zharkov To cite this version: Alexander Popov, Inga Grin, Antonina Dvornikova, Bakhyt Matkarimov, Regina Groisman, et al.. Reading targeted DNA damage in the active demethylation pathway: role of accessory domains of eukaryotic AP endonucleases and thymine-DNA glycosylases. Journal of Molecular Biology, Elsevier, 2020, 432 (6), pp.1747-1768. 10.1016/j.jmb.2019.12.020. hal-03060641 HAL Id: hal-03060641 https://hal.archives-ouvertes.fr/hal-03060641 Submitted on 14 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. *Manuscript Click here to view linked References Reading targeted DNA damage in the active demethylation 1 pathway: role of accessory domains of eukaryotic AP endonucleases 2 3 4 and thymine-DNA glycosylases 5 1 1,2 1,2 6 Alexander V. Popov , Inga R. Grin , Antonina P. Dvornikova -

DNA Translocases Rrp1 and Rrp2 Have Distinct
© 2020. Published by The Company of Biologists Ltd | Journal of Cell Science (2020) 133, jcs230193. doi:10.1242/jcs.230193 RESEARCH ARTICLE Schizosaccharomyces pombe DNA translocases Rrp1 and Rrp2 have distinct roles at centromeres and telomeres that ensure genome stability Anna Barg-Wojas1, Jakub Muraszko1,‡, Karol Kramarz2,‡, Kamila Schirmeisen1,*, Gabriela Baranowska1, Antony M. Carr3 and Dorota Dziadkowiec1,§ ABSTRACT a SWI2/SNF2 DNA translocase and SUMO-targeted ubiquitin The regulation of telomere and centromere structure and function is ligase (STUbL). essential for maintaining genome integrity. Schizosaccharomyces Telomeres and centromeres are potentially difficult to replicate pombe Rrp1 and Rrp2 are orthologues of Saccharomyces cerevisiae regions due to the presence of repetitive sequences that can form Uls1, a SWI2/SNF2 DNA translocase and SUMO-targeted ubiquitin secondary structures. These repetitive sequences are often unstable ligase. Here, we show that Rrp1 or Rrp2 overproduction leads to and constitute the hotspots of replication fork arrest and chromosome instability and growth defects, a reduction in global recombination. HR proteins act at arrested replication forks: histone levels and mislocalisation of centromere-specific histone Rad51 binding promotes the stability of the fork itself (Mizuno Cnp1. These phenotypes depend on putative DNA translocase et al., 2013; Schlacher et al., 2011), whereas, when the fork is activities of Rrp1 and Rrp2, suggesting that Rrp1 and Rrp2 may be inactivated, the strand exchange activity of Rad51 promotes the involved in modulating nucleosome dynamics. Furthermore, we reconstitution of replication (Lambert et al., 2005; McGlynn and confirm that Rrp2, but not Rrp1, acts at telomeres, reflecting a Lloyd, 2002). Indeed, S. pombe Rad51 localises to centromeres previously described interaction between Rrp2 and Top2. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Drosophila Rrpl Protein: an Apurinic Endonuclease with Homologous
Proc. NatI. Acad. Sci. USA Vol. 88, pp. 6780-6784, August 1991 Biochemistry Drosophila Rrpl protein: An apurinic endonuclease with homologous recombination activities (strand transfer/exodeoxyrlbonudease/DNA repair) MIRIAM SANDER*t, KY LOWENHAUPTt, AND ALEXANDER RICHt *Laboratory of Genetics D3-04, National Institute of Environmental Health Sciences, P.O. Box 12233, Research Triangle Park, NC 27709; and tDepartment of Biology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 Contributed by Alexander Rich, April 26, 1991 ABSTRACT A protein previously purified from Drosoph- proteins in the data base. Enzymatic characterization of the ia embryo extracts by a DNA strand transfer assay, Rrpl two bacterial enzymes suggests strongly that the homologous (recombination repair protein 1), has an N-terminal 427-amino domain is sufficient for AP endonuclease and 3' exonuclease acid region unrelated to known proteins, and a 252-amino acid activities (20, 21). The strand transfer activity of Rrpi has C-terminal region with sequence homology to two DNA repair been characterized (13). This activity copurifies with a poly- nucleases, Escherichia coli exonuclease HI and Streptococcus peptide of 105 kDa by electrophoretic mobility during SDS/ pneumoniae exonuclease A, which are known to be active as PAGE. Expression of the gene encoding this polypeptide in apurinic endonucleases and as double-stranded DNA 3' exo- E. coli has confirmed that it is active in strand transfer (M.S., nucleases. We demonstrate here that purified Rrpl has apu- unpublished results). In this report, we demonstrate that rnic endonuclease and double-stranded DNA 3' exonuclease Rrpl has associated AP endonuclease, 3' exonuclease, and activities and carries out singe-stranded DNA renaturation in single-stranded DNA (ssDNA) renaturation activities. -

Multiple Common Variants for Celiac Disease Influencing Immune Gene Expression
Europe PMC Funders Group Author Manuscript Nat Genet. Author manuscript; available in PMC 2010 October 01. Published in final edited form as: Nat Genet. 2010 April ; 42(4): 295–302. doi:10.1038/ng.543. Europe PMC Funders Author Manuscripts Multiple common variants for celiac disease influencing immune gene expression Patrick CA Dubois1,37, Gosia Trynka2,37, Lude Franke1,2, Karen A Hunt1, Jihane Romanos2, Alessandra Curtotti3, Alexandra Zhernakova4, Graham AR Heap1, Róza Ádány5, Arpo Aromaa6, Maria Teresa Bardella7,8, Leonard H van den Berg9, Nicholas A Bockett1, Emilio G. de la Concha10, Bárbara Dema10, Rudolf SN Fehrmann2, Miguel Fernández-Arquero10, Szilvia Fiatal5,11, Elvira Grandone12, Peter M Green13, Harry JM Groen14, Rhian Gwilliam15, Roderick HJ Houwen16, Sarah E Hunt15, Katri Kaukinen17, Dermot Kelleher18, Ilma Korponay-Szabo19,20, Kalle Kurppa17, Padraic MacMathuna21, Markku Mäki17, Maria Cristina Mazzilli22, Owen T McCann15, M Luisa Mearin23, Charles A Mein3, Muddassar M Mirza13, Vanisha Mistry1, Barbara Mora22, Katherine I Morley15, Chris J Mulder24, Joseph A Murray25, Concepción Núñez10, Elvira Oosterom2, Roel A Ophoff26,27, Isabel Polanco28, Leena Peltonen15,29, Mathieu Platteel2, Anna Rybak30, Veikko Salomaa6, Joachim J Schweizer23, Maria Pia Sperandeo31, Greetje J Tack24, Graham Turner18, Jan H Veldink9, Wieke HM Verbeek24, Rinse K Weersma2,32, Victorien M Wolters16, Elena Urcelay10, Bozena Cukrowska33, Luigi Greco31, Susan L. Neuhausen34, Ross McManus18, Donatella Barisani35, Panos Deloukas15, Jeffrey C Barrett15, Paivi -

The Biological Age of a Bloodstain Donor Author(S): Jack Ballantyne, Ph.D
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: The Biological Age of a Bloodstain Donor Author(s): Jack Ballantyne, Ph.D. Document Number: 251894 Date Received: July 2018 Award Number: 2009-DN-BX-K179 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. National Center for Forensic Science University of Central Florida P.O. Box 162367 · Orlando, FL 32826 Phone: 407.823.4041 Fax: 407.823.4042 Web site: http://www.ncfs.org/ Biological Evidence _________________________________________________________________________________________________________ The Biological Age of a Bloodstain Donor FINAL REPORT May 27, 2014 Department of Justice, National Institute of justice Award Number: 2009-DN-BX-K179 (1 October 2009 – 31 May 2014) _________________________________________________________________________________________________________ Principal Investigator: Jack Ballantyne, PhD Professor Department of Chemistry Associate Director for Research National Center for Forensic Science P.O. Box 162367 Orlando, FL 32816-2366 Phone: (407) 823 4440 Fax: (407) 823 4042 e-mail: [email protected] 1 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -

Differential Mechanisms of Tolerance to Extreme Environmental
www.nature.com/scientificreports OPEN Diferential mechanisms of tolerance to extreme environmental conditions in tardigrades Dido Carrero*, José G. Pérez-Silva , Víctor Quesada & Carlos López-Otín * Tardigrades, also known as water bears, are small aquatic animals that inhabit marine, fresh water or limno-terrestrial environments. While all tardigrades require surrounding water to grow and reproduce, species living in limno-terrestrial environments (e.g. Ramazzottius varieornatus) are able to undergo almost complete dehydration by entering an arrested state known as anhydrobiosis, which allows them to tolerate ionic radiation, extreme temperatures and intense pressure. Previous studies based on comparison of the genomes of R. varieornatus and Hypsibius dujardini - a less tolerant tardigrade - have pointed to potential mechanisms that may partially contribute to their remarkable ability to resist extreme physical conditions. In this work, we have further annotated the genomes of both tardigrades using a guided approach in search for novel mechanisms underlying the extremotolerance of R. varieornatus. We have found specifc amplifcations of several genes, including MRE11 and XPC, and numerous missense variants exclusive of R. varieornatus in CHEK1, POLK, UNG and TERT, all of them involved in important pathways for DNA repair and telomere maintenance. Taken collectively, these results point to genomic features that may contribute to the enhanced ability to resist extreme environmental conditions shown by R. varieornatus. Tardigrades are small -

Involvement of Schizosaccharomyces Pombe Rrp1 and Rrp2 in The
8196–8209 Nucleic Acids Research, 2013, Vol. 41, No. 17 Published online 4 July 2013 doi:10.1093/nar/gkt564 Involvement of Schizosaccharomyces pombe rrp1+ and rrp2+ in the Srs2- and Swi5/Sfr1-dependent pathway in response to DNA damage and replication inhibition Dorota Dziadkowiec1,*, Karol Kramarz1, Karolina Kanik1, Piotr Wis´ niewski2 and Antony M. Carr3 1Faculty of Biotechnology, Wrocław University, Przybyszewskiego 63-77, 51-148 Wrocław, Poland, 2Institute of Low Temperature and Structure Research, Polish Academy of Sciences, PO Box 1410, 50-950 Wrocław, Downloaded from Poland and 3Genome Damage and Stability Centre, University of Sussex, Falmer, Brighton, BN1 9RQ, UK Received April 15, 2013; Revised June 1, 2013; Accepted June 4, 2013 ABSTRACT INTRODUCTION http://nar.oxfordjournals.org/ Previously we identified Rrp1 and Rrp2 as two In all organisms, homologous recombination (HR) is a proteins required for the Sfr1/Swi5-dependent high-fidelity DNA repair pathway, essential for the branch of homologous recombination (HR) in repair of DNA double-strand breaks (DSBs) and for Schizosaccharomyces pombe. Here we use a yeast normal DNA replication. The RecA/Rad51 family of proteins forms filaments on single-stranded DNA two-hybrid approach to demonstrate that Rrp1 and (ssDNA), which catalyze homology search DNA strand Rrp2 can interact with each other and with Swi5, an invasion reactions, the hallmark of HR (1). HR mediator protein. Rrp1 and Rrp2 form co- Nucleofilament formation by Rad51 recombinase at Sussex Language Institute on September 10, 2014 localizing methyl methanesulphonate–induced foci (formerly Rhp51 in Schizosaccharomyces pombe) (2) is in nuclei, further suggesting they function as a assisted by a group of proteins called recombination me- complex. -

S. Pombe DNA Translocases Rrp1 and Rrp2 Have Distinct Roles At
bioRxiv preprint doi: https://doi.org/10.1101/738435; this version posted August 16, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. S. pombe DNA translocases Rrp1 and Rrp2 have distinct roles at centromeres and telomeres that ensure genome stability Anna Barg-Wojas1, Kamila Schirmeisen1,4, Jakub Muraszko1, Karol Kramarz2, Gabriela Baranowska1, Antony M. Carr3, Dorota Dziadkowiec1,* 1 Faculty of Biotechnology, University of Wrocław, Poland 2 Institut Curie, Centre National de la Recherche Scientifique, Orsay, France 3 Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, BN1 9RQ, United Kingdom 4 present address: Institut Curie, Centre National de la Recherche Scientifique, Orsay, France * corresponding author: [email protected] Key words: nucleosome dynamics; DNA translocase; centromere and telomere structure SUMMARY STATEMENT Schizosaccharomyces pombe DNA translocases Rrp1 and Rrp2 modulate centromere and telomere maintenance pathways and dysregulation of their activity leads to genome instability. ABSTRACT Homologous recombination (HR) is a DNA repair mechanism that ensures, together with heterochromatin machinery, the proper replication, structure and function of telomeres and centromeres that is essential for the maintenance of genome integrity. Schizosaccharomyces pombe Rrp1 and Rrp2 participate in HR and are orthologues of Saccharomyces cerevisiae Uls1, a SWI2/SNF2 DNA translocase and SUMO-Targeted Ubiquitin Ligase. We show that Rrp1 or Rrp2 upregulation leads to chromosome instability and growth defects. These phenotypes depend on putative DNA translocase activities of Rrp1 and Rrp2. Either Rrp1 or Rrp2 overproduction results in a reduction in global histone levels, suggesting that Rrp1 and Rrp2 may modulate nucleosome dynamics. -

Systematic Proteome and Proteostasis Profiling in Human Trisomy
ARTICLE DOI: 10.1038/s41467-017-01422-6 OPEN Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells Yansheng Liu 1, Christelle Borel2,LiLi3, Torsten Müller1, Evan G. Williams1, Pierre-Luc Germain4, Marija Buljan1, Tatjana Sajic1, Paul J. Boersema5, Wenguang Shao1, Marco Faini1, Giuseppe Testa4,6, Andreas Beyer 3, Stylianos E. Antonarakis2,7 & Ruedi Aebersold 1,8 Down syndrome (DS) is mostly caused by a trisomy of the entire Chromosome 21 (Trisomy 21, T21). Here, we use SWATH mass spectrometry to quantify protein abundance and protein turnover in fibroblasts from a monozygotic twin pair discordant for T21, and to profile protein expression in 11 unrelated DS individuals and matched controls. The integration of the steady- state and turnover proteomic data indicates that protein-specific degradation of members of stoichiometric complexes is a major determinant of T21 gene dosage outcome, both within and between individuals. This effect is not apparent from genomic and transcriptomic data. The data also reveal that T21 results in extensive proteome remodeling, affecting proteins encoded by all chromosomes. Finally, we find broad, organelle-specific post-transcriptional effects such as significant downregulation of the mitochondrial proteome contributing to T21 hallmarks. Overall, we provide a valuable proteomic resource to understand the origin of DS phenotypic manifestations. 1 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland. 2 Department of Genetic Medicine and Development, University of Geneva Medical School, and University Hospitals of Geneva, 1211 Geneva, Switzerland. 3 Cellular Networks and Systems Biology, University of Cologne, CECAD, University of Cologne, 50931 Cologne, Germany. 4 Department of Experimental Oncology, European Institute of Oncology, 20139 Milan, Italy.