Downloaded from Bioscientifica.Com at 09/24/2021 09:36:43AM Via Free Access Review a GEORGES and Others Forkhead Box L2 in the Ovary 52:1 R18
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Transcription Factors SOHLH1 and SOHLH2 Coordinate Oocyte Differentiation Without Affecting Meiosis I
Transcription factors SOHLH1 and SOHLH2 coordinate oocyte differentiation without affecting meiosis I Yong-Hyun Shin, … , Vasil Mico, Aleksandar Rajkovic J Clin Invest. 2017;127(6):2106-2117. https://doi.org/10.1172/JCI90281. Research Article Development Reproductive biology Following migration of primordial germ cells to the genital ridge, oogonia undergo several rounds of mitotic division and enter meiosis at approximately E13.5. Most oocytes arrest in the dictyate (diplotene) stage of meiosis circa E18.5. The genes necessary to drive oocyte differentiation in parallel with meiosis are unknown. Here, we have investigated whether expression of spermatogenesis and oogenesis bHLH transcription factor 1 (Sohlh1) and Sohlh2 coordinates oocyte differentiation within the embryonic ovary. We found that SOHLH2 protein was expressed in the mouse germline as early as E12.5 and preceded SOHLH1 protein expression, which occurred circa E15.5. SOHLH1 protein appearance at E15.5 correlated with SOHLH2 translocation from the cytoplasm into the nucleus and was dependent on SOHLH1 expression. NOBOX oogenesis homeobox (NOBOX) and LIM homeobox protein 8 (LHX8), two important regulators of postnatal oogenesis, were coexpressed with SOHLH1. Single deficiency of Sohlh1 or Sohlh2 disrupted the expression of LHX8 and NOBOX in the embryonic gonad without affecting meiosis. Sohlh1-KO infertility was rescued by conditional expression of the Sohlh1 transgene after the onset of meiosis. However, Sohlh1 or Sohlh2 transgene expression could not rescue Sohlh2-KO infertility due to a lack of Sohlh1 or Sohlh2 expression in rescued mice. Our results indicate that Sohlh1 and Sohlh2 are essential regulators of oocyte differentiation but do not affect meiosis I. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Dlx3b/4B Is Required for Early-Born but Not Later-Forming Sensory Hair Cells
© 2017. Published by The Company of Biologists Ltd | Biology Open (2017) 6, 1270-1278 doi:10.1242/bio.026211 RESEARCH ARTICLE Dlx3b/4b is required for early-born but not later-forming sensory hair cells during zebrafish inner ear development Simone Schwarzer, Sandra Spieß, Michael Brand and Stefan Hans* ABSTRACT complex arrangement of mechanosensory hair cells, nonsensory Morpholino-mediated knockdown has shown that the homeodomain supporting cells and sensory neurons (Barald and Kelley, 2004; Raft transcription factors Dlx3b and Dlx4b are essential for proper and Groves, 2015; Whitfield, 2015). Inner ear formation is a induction of the otic-epibranchial progenitor domain (OEPD), as multistep process initiated by the establishment of the preplacodal well as subsequent formation of sensory hair cells in the developing region, a zone of ectoderm running around the anterior border of the zebrafish inner ear. However, increasing use of reverse genetic neural plate containing precursors for all sensory placodes (Streit, approaches has revealed poor correlation between morpholino- 2007). The preplacodal region is further specified into a common induced and mutant phenotypes. Using CRISPR/Cas9-mediated otic-epibranchial progenitor domain (OEPD) that in zebrafish also mutagenesis, we generated a defined deletion eliminating the entire contains the progenitors of the anterior lateral line ganglion (Chen open reading frames of dlx3b and dlx4b (dlx3b/4b) and investigated a and Streit, 2013; McCarroll et al., 2012; Hans et al., 2013). potential phenotypic -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

An Integrative, Genomic, Transcriptomic and Network-Assisted
Yan et al. BMC Medical Genomics 2020, 13(Suppl 5):39 https://doi.org/10.1186/s12920-020-0675-4 RESEARCH Open Access An integrative, genomic, transcriptomic and network-assisted study to identify genes associated with human cleft lip with or without cleft palate Fangfang Yan1†, Yulin Dai1†, Junichi Iwata2,3, Zhongming Zhao1,4,5* and Peilin Jia1* From The International Conference on Intelligent Biology and Medicine (ICIBM) 2019 Columbus, OH, USA. 9-11 June 2019 Abstract Background: Cleft lip with or without cleft palate (CL/P) is one of the most common congenital human birth defects. A combination of genetic and epidemiology studies has contributed to a better knowledge of CL/P-associated candidate genes and environmental risk factors. However, the etiology of CL/P remains not fully understood. In this study, to identify new CL/P-associated genes, we conducted an integrative analysis using our in-house network tools, dmGWAS [dense module search for Genome-Wide Association Studies (GWAS)] and EW_dmGWAS (Edge-Weighted dmGWAS), in a combination with GWAS data, the human protein-protein interaction (PPI) network, and differential gene expression profiles. Results: A total of 87 genes were consistently detected in both European and Asian ancestries in dmGWAS. There were 31.0% (27/87) showed nominal significance with CL/P (gene-based p < 0.05), with three genes showing strong association signals, including KIAA1598, GPR183,andZMYND11 (p <1×10− 3). In EW_dmGWAS, we identified 253 and 245 module genes associated with CL/P for European ancestry and the Asian ancestry, respectively. Functional enrichment analysis demonstrated that these genes were involved in cell adhesion, protein localization to the plasma membrane, the regulation of the apoptotic signaling pathway, and other pathological conditions. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Supplementary Table 5. Clover Results Indicate the Number Of
Supplementary Table 5. Clover results indicate the number of chromosomes with transcription factor binding motifs statistically over‐ or under‐represented in HTE DHS within intergenic sequence (more than 2kb outside of any gene). Analysis was divided into three groups (all DHS, HTE‐selective DHS, and ubiquitous DHS). Motifs with more than one entry in the databases utilized were edited to retain only the first occurrence of the motif. All DHS x Intergenic TEselective DHS x Intergenic Ubiquitous DHS x Intergenic ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 MA0002.2 RUNX1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0003.1 TFAP2A 23 0 MA0089.1 NFE2L1::MafG 23 0 MA0068.1 Pax4 23 0 MA0039.2 Klf4 23 0 MA0098.1 ETS1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0099.2 AP1 23 0 MA0098.1 ETS1 23 0 MA0056.1 MZF1_1‐4 23 0 MA0136.1 ELF5 23 0 MA0139.1 CTCF 23 0 MA0079.2 SP1 23 0 MA0145.1 Tcfcp2l1 23 0 V$ALX3_01 ALX‐3 23 0 MA0080.2 SPI1 23 0 MA0150.1 NFE2L2 23 0 V$ALX4_02 Alx‐4 23 0 myocyte enhancer MA0081.1 SPIB 23 0 MA0156.1 FEV 23 0 V$AMEF2_Q6 factor 23 0 MA0089.1 NFE2L1::MafG 23 0 V$AP1FJ_Q2 activator protein 1 23 0 V$AP1_01 AP‐1 23 0 MA0090.1 TEAD1 23 0 V$AP4_Q5 activator protein 4 23 0 V$AP2_Q6_01 AP‐2 23 0 MA0098.1 ETS1 23 0 V$AR_Q6 half‐site matrix 23 0 V$ARX_01 Arx 23 0 BTB and CNC homolog MA0099.2 AP1 23 0 V$BACH1_01 1 23 0 V$BARHL1_01 Barhl‐1 23 0 BTB and CNC homolog MA0136.1 ELF5 23 0 V$BACH2_01 2 23 0 V$BARHL2_01 Barhl2 23 0 MA0139.1 CTCF 23 0 V$CMAF_02 C‐MAF 23 0 V$BARX1_01 Barx1 23 0 MA0144.1 Stat3 23 0 -
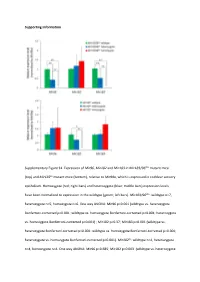
Supporting Information Supplementary Figure S1
Supporting Information Supplementary Figure S1. Expression of Mir96, Mir182 and Mir183 in Mir183/96dko mutant mice (top) and Mir182ko mutant mice (bottom), relative to Mir99a, which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96dko: wildtype n=7, heterozygote n=5, homozygote n=6. One way ANOVA: Mir96 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p<0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p=0.001) ; Mir182 p=0.37; Mir183 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p=0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p<0.001). Mir182ko: wildtype n=4, heterozygote n=4, homozygote n=4. One way ANOVA: Mir96 p=0.685; Mir182 p=0.003 (wildtype vs. heterozygote Bonferroni‐corrected p=0.397; wildtype vs. homozygote Bonferroni‐corrected p=0.003; heterozygote vs. homozygote Bonferroni‐corrected p=0.032); Mir183 p=0.04 (wildtype vs. heterozygote Bonferroni‐corrected p=1.0; wildtype vs. homozygote Bonferroni‐corrected p=0.068; heterozygote vs. homozygote Bonferroni‐corrected p=0.094), Error bars are standard deviation (* = P < 0.05, ** = P ≤ 0.01). Supplementary Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. Supplementary Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182ko mice at all ages tested. -

Supplement. Transcriptional Factors (TF), Protein Name and Their Description Or Function
Supplement. Transcriptional factors (TF), protein name and their description or function. TF Protein name TF description/function ARID3A AT rich interactive domain 3A (BRIGHT-like) This gene encodes a member of the ARID (AT-rich interaction domain) family of DNA binding proteins. ATF4 Activating Transcription Factor 4 Transcriptional activator. Binds the cAMP response element (CRE) (consensus: 5-GTGACGT[AC][AG]-3), a sequence present in many viral and cellular promoters. CTCF CCCTC-Binding Factor Chromatin binding factor that binds to DNA sequence specific sites. Involved in transcriptional regulation by binding to chromatin insulators and preventing interaction between promoter and nearby enhancers and silencers. The protein can bind a histone acetyltransferase (HAT)-containing complex and function as a transcriptional activator or bind a histone deacetylase (HDAC)-containing complex and function as a transcriptional repressor. E2F1-6 E2F transcription factors 1-6 The protein encoded by this gene is a member of the E2F family of transcription factors. The E2F family plays a crucial role in the control of cell cycle and action of tumor suppressor proteins and is also a target of the transforming proteins of small DNA tumor viruses. The E2F proteins contain several evolutionally conserved domains found in most members of the family. These domains include a DNA binding domain, a dimerization domain which determines interaction with the differentiation regulated transcription factor proteins (DP), a transactivation domain enriched in acidic amino acids, and a tumor suppressor protein association domain which is embedded within the transactivation domain. EBF1 Transcription factor COE1 EBF1 has been shown to interact with ZNF423 and CREB binding proteins. -

The Genetics of Non-Syndromic Primary Ovarian Insufficiency: a Systematic Review
Systematic Review The Genetics of Non-Syndromic Primary Ovarian Insufficiency: A Systematic Review Roberta Venturella, M.D.1, Valentino De Vivo, M.D.2, Annunziata Carlea, M.D.2, Pietro D’Alessandro, M.D.2, Gabriele Saccone, M.D.2*, Bruno Arduino, M.D.2, Francesco Paolo Improda, M.D.2, Daniela Lico, M.D.1, Erika Rania, M.D.1, Carmela De Marco, M.D.3, Giuseppe Viglietto, M.D.3, Fulvio Zullo, M.D.1 1. Department of Obstetrics and Gynaecology, Magna Graecia University of Catanzaro, Catanzaro, Italy 2. Department of Neuroscience, Reproductive Sciences and Dentistry, School of Medicine, University of Naples Federico II, Naples, Italy 3. Department of Experimental and Clinical Medicine, Magna Graecia University of Catanzaro, Catanzaro, Italy Abstract Several causes for primary ovarian insufficiency (POI) have been described, including iatrogenic and environmental factor, viral infections, chronic disease as well as genetic alterations. The aim of this review was to collect all the ge- netic mutations associated with non-syndromic POI. All studies, including gene screening, genome-wide study and as- sessing genetic mutations associated with POI, were included and analyzed in this systematic review. Syndromic POI and chromosomal abnormalities were not evaluated. Single gene perturbations, including genes on the X chromosome (such as BMP15, PGRMC1 and FMR1) and genes on autosomal chromosomes (such as GDF9, FIGLA, NOBOX, ESR1, FSHR and NANOS3) have a positive correlation with non-syndromic POI. Future strategies include linkage analysis of families with multiple affected members, array comparative genomic hybridization (CGH) for analysis of copy number variations, next generation sequencing technology and genome-wide data analysis.