Transcriptomic Profiling Identifies Differentially Expressed Genes In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Integrated and Functional Genomic Approaches to Elucidate Differential Genetic Dependencies in Melanoma
Integrated and Functional Genomic Approaches to Elucidate Differential Genetic Dependencies in Melanoma The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wong, Terence. 2018. Integrated and Functional Genomic Approaches to Elucidate Differential Genetic Dependencies in Melanoma. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42014990 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Integrated and Functional Genomic Approaches to Elucidate Differential Genetic Dependencies in Melanoma A dissertation presented by Terence Cheng Wong to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biological and Biomedical Sciences Harvard University Cambridge, Massachusetts November 2017 © 2017 Terence Cheng Wong All rights reserved. Dissertation Advisor: Levi Garraway Terence Cheng Wong Integrated and Functional Genomic Approaches to Elucidate Differential Genetic Dependencies in Melanoma ABSTRACT Genomic characterization of human cancers over the past decade has generated comprehensive catalogues of genetic alterations in cancer genomes. Many of these genetic events result in molecular or cellular changes that drive cancer cell phenotypes. In melanoma, a majority of tumors harbor mutations in the BRAF gene, leading to activation of the MAPK pathway and tumor initiation. The development and use of drugs that target the mutant BRAF protein and the MAPK pathway have produced significant clinical benefit in melanoma patients. -

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -

Dlx3b/4B Is Required for Early-Born but Not Later-Forming Sensory Hair Cells
© 2017. Published by The Company of Biologists Ltd | Biology Open (2017) 6, 1270-1278 doi:10.1242/bio.026211 RESEARCH ARTICLE Dlx3b/4b is required for early-born but not later-forming sensory hair cells during zebrafish inner ear development Simone Schwarzer, Sandra Spieß, Michael Brand and Stefan Hans* ABSTRACT complex arrangement of mechanosensory hair cells, nonsensory Morpholino-mediated knockdown has shown that the homeodomain supporting cells and sensory neurons (Barald and Kelley, 2004; Raft transcription factors Dlx3b and Dlx4b are essential for proper and Groves, 2015; Whitfield, 2015). Inner ear formation is a induction of the otic-epibranchial progenitor domain (OEPD), as multistep process initiated by the establishment of the preplacodal well as subsequent formation of sensory hair cells in the developing region, a zone of ectoderm running around the anterior border of the zebrafish inner ear. However, increasing use of reverse genetic neural plate containing precursors for all sensory placodes (Streit, approaches has revealed poor correlation between morpholino- 2007). The preplacodal region is further specified into a common induced and mutant phenotypes. Using CRISPR/Cas9-mediated otic-epibranchial progenitor domain (OEPD) that in zebrafish also mutagenesis, we generated a defined deletion eliminating the entire contains the progenitors of the anterior lateral line ganglion (Chen open reading frames of dlx3b and dlx4b (dlx3b/4b) and investigated a and Streit, 2013; McCarroll et al., 2012; Hans et al., 2013). potential phenotypic -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

FOXK Transcription Factors Regulation and Critical Role in Cancer
Cancer Letters 458 (2019) 1–12 Contents lists available at ScienceDirect Cancer Letters journal homepage: www.elsevier.com/locate/canlet Mini-review FOXK transcription factors: Regulation and critical role in cancer T Ying Liua, Wei Dingb,HuGea,d, Murugavel Ponnusamya, Qiong Wangd, Xiaodan Haoa, Wei Wua, ∗∗ ∗ Yuan Zhanga, Wanpeng Yua, Xiang Aoa, , Jianxun Wanga,c, a Institute for Translational Medicine, College of Medicine, Qingdao University, Qingdao 266021, China b Department of Comprehensive Internal Medicine, Affiliated Hospital, Qingdao University, Qingdao 266003, China c School of Basic Medical Sciences, Qingdao University, Qingdao 266071, China d Molecular Informatics Department, Hengrui Pharmaceutical Co., Ltd., Shanghai 200245, China ARTICLE INFO ABSTRACT Keywords: Growing evidence suggests that alterations of gene expression including expression and activities of transcription FOXK1 factors are closely associated with carcinogenesis. Forkhead Box Class K (FOXK) proteins, FOXK1 and FOXK2, FOXK2 are a family of evolutionarily conserved transcriptional factors, which have recently been recognized as key ncRNAs transcriptional regulators involved in many types of cancer. Members of the FOXK family mediate a wide Biomarker spectrum of biological processes, including cell proliferation, differentiation, apoptosis, autophagy, cell cycle Therapeutic target progression, DNA damage and tumorigenesis. Therefore, the deregulation of FOXKs can affect the cell fate and they promote tumorigenesis as well as cancer progression. The mechanisms of FOXKs regulation including post- translational modifications (PTMs), microRNAs (miRNAs) and protein–protein interactions are well demon- strated. However, the detailed mechanisms of FOXKs activation and deregulation in cancer progression are still inconclusive. In this review, we summarize the regulatory mechanisms of FOXKs expression and activity, and their role in the development and progression of cancer. -

Supplementary Table 5. Clover Results Indicate the Number Of
Supplementary Table 5. Clover results indicate the number of chromosomes with transcription factor binding motifs statistically over‐ or under‐represented in HTE DHS within intergenic sequence (more than 2kb outside of any gene). Analysis was divided into three groups (all DHS, HTE‐selective DHS, and ubiquitous DHS). Motifs with more than one entry in the databases utilized were edited to retain only the first occurrence of the motif. All DHS x Intergenic TEselective DHS x Intergenic Ubiquitous DHS x Intergenic ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 ID Name p < 0.01 p > 0.99 MA0002.2 RUNX1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0003.1 TFAP2A 23 0 MA0089.1 NFE2L1::MafG 23 0 MA0068.1 Pax4 23 0 MA0039.2 Klf4 23 0 MA0098.1 ETS1 23 0 MA0080.2 SPI1 23 0 MA0055.1 Myf 23 0 MA0099.2 AP1 23 0 MA0098.1 ETS1 23 0 MA0056.1 MZF1_1‐4 23 0 MA0136.1 ELF5 23 0 MA0139.1 CTCF 23 0 MA0079.2 SP1 23 0 MA0145.1 Tcfcp2l1 23 0 V$ALX3_01 ALX‐3 23 0 MA0080.2 SPI1 23 0 MA0150.1 NFE2L2 23 0 V$ALX4_02 Alx‐4 23 0 myocyte enhancer MA0081.1 SPIB 23 0 MA0156.1 FEV 23 0 V$AMEF2_Q6 factor 23 0 MA0089.1 NFE2L1::MafG 23 0 V$AP1FJ_Q2 activator protein 1 23 0 V$AP1_01 AP‐1 23 0 MA0090.1 TEAD1 23 0 V$AP4_Q5 activator protein 4 23 0 V$AP2_Q6_01 AP‐2 23 0 MA0098.1 ETS1 23 0 V$AR_Q6 half‐site matrix 23 0 V$ARX_01 Arx 23 0 BTB and CNC homolog MA0099.2 AP1 23 0 V$BACH1_01 1 23 0 V$BARHL1_01 Barhl‐1 23 0 BTB and CNC homolog MA0136.1 ELF5 23 0 V$BACH2_01 2 23 0 V$BARHL2_01 Barhl2 23 0 MA0139.1 CTCF 23 0 V$CMAF_02 C‐MAF 23 0 V$BARX1_01 Barx1 23 0 MA0144.1 Stat3 23 0 -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -
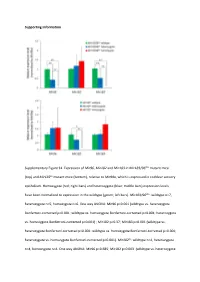
Supporting Information Supplementary Figure S1
Supporting Information Supplementary Figure S1. Expression of Mir96, Mir182 and Mir183 in Mir183/96dko mutant mice (top) and Mir182ko mutant mice (bottom), relative to Mir99a, which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96dko: wildtype n=7, heterozygote n=5, homozygote n=6. One way ANOVA: Mir96 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p<0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p=0.001) ; Mir182 p=0.37; Mir183 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p=0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p<0.001). Mir182ko: wildtype n=4, heterozygote n=4, homozygote n=4. One way ANOVA: Mir96 p=0.685; Mir182 p=0.003 (wildtype vs. heterozygote Bonferroni‐corrected p=0.397; wildtype vs. homozygote Bonferroni‐corrected p=0.003; heterozygote vs. homozygote Bonferroni‐corrected p=0.032); Mir183 p=0.04 (wildtype vs. heterozygote Bonferroni‐corrected p=1.0; wildtype vs. homozygote Bonferroni‐corrected p=0.068; heterozygote vs. homozygote Bonferroni‐corrected p=0.094), Error bars are standard deviation (* = P < 0.05, ** = P ≤ 0.01). Supplementary Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. Supplementary Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182ko mice at all ages tested. -

Supplement. Transcriptional Factors (TF), Protein Name and Their Description Or Function
Supplement. Transcriptional factors (TF), protein name and their description or function. TF Protein name TF description/function ARID3A AT rich interactive domain 3A (BRIGHT-like) This gene encodes a member of the ARID (AT-rich interaction domain) family of DNA binding proteins. ATF4 Activating Transcription Factor 4 Transcriptional activator. Binds the cAMP response element (CRE) (consensus: 5-GTGACGT[AC][AG]-3), a sequence present in many viral and cellular promoters. CTCF CCCTC-Binding Factor Chromatin binding factor that binds to DNA sequence specific sites. Involved in transcriptional regulation by binding to chromatin insulators and preventing interaction between promoter and nearby enhancers and silencers. The protein can bind a histone acetyltransferase (HAT)-containing complex and function as a transcriptional activator or bind a histone deacetylase (HDAC)-containing complex and function as a transcriptional repressor. E2F1-6 E2F transcription factors 1-6 The protein encoded by this gene is a member of the E2F family of transcription factors. The E2F family plays a crucial role in the control of cell cycle and action of tumor suppressor proteins and is also a target of the transforming proteins of small DNA tumor viruses. The E2F proteins contain several evolutionally conserved domains found in most members of the family. These domains include a DNA binding domain, a dimerization domain which determines interaction with the differentiation regulated transcription factor proteins (DP), a transactivation domain enriched in acidic amino acids, and a tumor suppressor protein association domain which is embedded within the transactivation domain. EBF1 Transcription factor COE1 EBF1 has been shown to interact with ZNF423 and CREB binding proteins. -

FOXK1 Promotes Malignant Progression of Breast Cancer by Activating PI3K/AKT/Mtor Signaling Pathway
European Review for Medical and Pharmacological Sciences 2019; 23: 9978-9987 FOXK1 promotes malignant progression of breast cancer by activating PI3K/AKT/mTOR signaling pathway Z.-Q. LI1, M. QU2, H.-X. WAN1, H. WANG3, Q. DENG4, Y. ZHANG5 1Department of Oncology, Sanya People’s Hospital, Sanya, China 2Special Care Unit, Sanya People’s Hospital, Sanya, China 3Department of Pathology, Sanya People’s Hospital, Sanya, China 4Department of Surgical Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China 5Department of Ultrasound in Medicine, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China Zhiqiang Li and Miao Qu contributed equally to this work Abstract. – OBJECTIVE: The aim of this the PI3K/AKT/mTOR signaling pathway, thereby study was to investigate the expression charac- promoting the malignant progression of BCa. teristics of forkhead box K1 (FOXK1) in breast Finally, PI3Kα/mTOR-IN-1, which was the inhib- cancer (BCa). Meanwhile, its relationship with itor of the PI3K/AKT/mTOR signaling pathway, clinicopathology and prognosis of patients with significantly reversed the proliferative capacity BCa was also explored. of cells in FOXK1 overexpression group, as well PATIENTS AND METHODS: The expression as enhanced anti-apoptotic ability. level of FOXK1 in 65 paired BCa tissues and pa- CONCLUSIONS: FOXK1 expression was re- ra-cancerous tissues was detected by quan- markably increased both in BCa tissues and titative Real Time-Polymerase Chain Reaction cells. Meanwhile, it was markedly associated (qRT-PCR). The relationship between FOXK1 ex- with pathological stage and poor prognosis of pression and BCa pathological parameters as patients. Besides, FOXK1 might promote the well as the prognosis of patients was analyzed.