Genome-Guided Insights Reveal Organophosphate- Degrading
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Sphingopyxis Italica, Sp. Nov., Isolated from Roman Catacombs 1 2
View metadata, citation and similar papers at core.ac.uk brought to you by CORE IJSEM Papers in Press. Published December 21, 2012 as doi:10.1099/ijs.0.046573-0 provided by Digital.CSIC 1 Sphingopyxis italica, sp. nov., isolated from Roman catacombs 2 3 Cynthia Alias-Villegasª, Valme Jurado*ª, Leonila Laiz, Cesareo Saiz-Jimenez 4 5 Instituto de Recursos Naturales y Agrobiologia, IRNAS-CSIC, 6 Apartado 1052, 41080 Sevilla, Spain 7 8 * Corresponding author: 9 Valme Jurado 10 Instituto de Recursos Naturales y Agrobiologia, IRNAS-CSIC 11 Apartado 1052, 41080 Sevilla, Spain 12 Tel. +34 95 462 4711, Fax +34 95 462 4002 13 E-mail: [email protected] 14 15 ª These authors contributed equally to this work. 16 17 Keywords: Sphingopyxis italica, Roman catacombs, rRNA, sequence 18 19 The sequence of the 16S rRNA gene from strain SC13E-S71T can be accessed 20 at Genbank, accession number HE648058. 21 22 A Gram-negative, aerobic, motile, rod-shaped bacterium, strain SC13E- 23 S71T, was isolated from tuff, the volcanic rock where was excavated the 24 Roman Catacombs of Saint Callixtus in Rome, Italy. Analysis of 16S 25 rRNA gene sequences revealed that strain SC13E-S71T belongs to the 26 genus Sphingopyxis, and that it shows the greatest sequence similarity 27 with Sphingopyxis chilensis DSMZ 14889T (98.72%), Sphingopyxis 28 taejonensis DSMZ 15583T (98.65%), Sphingopyxis ginsengisoli LMG 29 23390T (98.16%), Sphingopyxis panaciterrae KCTC12580T (98.09%), 30 Sphingopyxis alaskensis DSM 13593T (98.09%), Sphingopyxis 31 witflariensis DSM 14551T (98.09%), Sphingopyxis bauzanensis DSM 32 22271T (98.02%), Sphingopyxis granuli KCTC12209T (97.73%), 33 Sphingopyxis macrogoltabida KACC 10927T (97.49%), Sphingopyxis 34 ummariensis DSM 24316T (97.37%) and Sphingopyxis panaciterrulae T 35 KCTC 22112 (97.09%). -

Sphingopyxis Soli Sp. Nov., Isolated from Landfill Soil
International Journal of Systematic and Evolutionary Microbiology (2010), 60, 1682–1686 DOI 10.1099/ijs.0.013128-0 Sphingopyxis soli sp. nov., isolated from landfill soil Jung-Hye Choi,1 Min-Soo Kim,1,2 Mi-Ja Jung,1 Seong Woon Roh,1,2 Kee-Sun Shin2 and Jin-Woo Bae1 Correspondence 1Department of Life and Nanopharmaceutical Sciences and Department of Biology, Jin-Woo Bae Kyung Hee University, Seoul 130-701, Republic of Korea [email protected] 2University of Science and Technology, BRC, KRIBB, Daejeon 305-333, Republic of Korea A Gram-negative, aerobic, rod-shaped, motile, oxidase-positive, catalase-negative bacterium, designated strain BL03T, was isolated from landfill soil in Pohang, Republic of Korea. Colonies on Luria–Bertani agar plates were yellow. The strain grew in the presence of 0–3 % (w/v) NaCl, at 15–42 6C and at pH 5.0–9.5. The predominant ubiquinone was Q-10, and the major cellular fatty acids were C17 : 1v6c,C15 : 0 2-OH and C18 : 1v7c. Polar lipids detected were phosphatidylmonomethylethanolamine, diphosphatidylglycerol, phosphatidylcholine, phosphatidylethanolamine, phosphatidylglycerol, sphingoglycolipid and an unknown glycolipid. Spermidine was identified as the major polyamine component. Phylogenetic analysis based on 16S rRNA gene sequences showed that strain BL03T belongs to the genus Sphingopyxis with high sequence similarity to Sphingopyxis taejonensis JSS54T (97.8 %), Sphingopyxis alaskensis RB2256T (97.4 %) and Sphingopyxis chilensis S37T (96.9 %). Levels of DNA–DNA relatedness between strain BL03T and the above three type strains were only 10.3–40.3 %. The DNA G+C content of strain BL03T was 65.9 mol%. -

Sphingopyxis Sp. Strain OPL5, an Isoprene-Degrading Bacterium from the Sphingomonadaceae Family Isolated from Oil Palm Leaves
microorganisms Article Sphingopyxis sp. Strain OPL5, an Isoprene-Degrading Bacterium from the Sphingomonadaceae Family Isolated from Oil Palm Leaves Nasmille L. Larke-Mejía 1 , Ornella Carrión 1 , Andrew T. Crombie 2, Terry J. McGenity 3 and J. Colin Murrell 1,* 1 School of Environmental Sciences, University of East Anglia, Norwich NR4 7TJ, UK; [email protected] (N.L.L.-M.); [email protected] (O.C.) 2 School of Biological Sciences, University of East Anglia, Norwich NR4 7TJ, UK; [email protected] 3 School of Life Sciences, University of Essex, Colchester CO4 3SQ, UK; [email protected] * Correspondence: [email protected]; Tel.: +44-01603-592959 Received: 2 September 2020; Accepted: 7 October 2020; Published: 10 October 2020 Abstract: The volatile secondary metabolite, isoprene, is released by trees to the atmosphere in enormous quantities, where it has important effects on air quality and climate. Oil palm trees, one of the highest isoprene emitters, are increasingly dominating agroforestry over large areas of Asia, with associated uncertainties over their effects on climate. Microbes capable of using isoprene as a source of carbon for growth have been identified in soils and in the tree phyllosphere, and most are members of the Actinobacteria. Here, we used DNA stable isotope probing to identify the isoprene-degrading bacteria associated with oil palm leaves and inhabiting the surrounding soil. Among the most abundant isoprene degraders of the leaf-associated community were members of the Sphingomonadales, although no representatives of this order were previously known to degrade isoprene. Informed by these data, we obtained representatives of the most abundant isoprene degraders in enrichments, including Sphingopyxis strain OPL5 (Sphingomonadales), able to grow on isoprene as the sole source of carbon and energy. -

Sphingopyxis Chilensis Sp. Nov., a Chlorophenol-Degrading Bacterium
International Journal of Systematic and Evolutionary Microbiology (2003), 53, 473–477 DOI 10.1099/ijs.0.02375-0 Note Sphingopyxis chilensis sp. nov., a chlorophenol-degrading bacterium that accumulates polyhydroxyalkanoate, and transfer of Sphingomonas alaskensis to Sphingopyxis alaskensis comb. nov. F. Godoy,1 M. Vancanneyt,2 M. Martı´nez,1 A. Steinbu¨chel,3 J. Swings2 and B. H. A. Rehm3 Correspondence 1Departamento de Microbiologı´a, Facultad de Ciencias Biolo´gicas, Universidad de Concepcio´n, B. H. A. Rehm Casilla 160-C Concepcio´n, Chile [email protected] 2BCCM/LMG Bacteria Collection, University of Ghent, K. L. Ledeganckstraat 35, B-9000 Gent, Belgium 3Institut fu¨r Mikrobiologie der Westfa¨lischen, Wilhelms–Universita¨t Mu¨nster, Corrensstrasse 3, D-48149 Mu¨nster, Germany The taxonomic position of a chlorophenol-degrading bacterium, strain S37T, was investigated. The 16S rDNA sequence indicated that this strain belongs to the genus Sphingopyxis, exhibiting high sequence similarity to the 16S rDNA sequences of Sphingomonas alaskensis LMG 18877T (98?8 %), Sphingopyxis macrogoltabida LMG 17324T (98?2 %), Sphingopyxis terrae IFO 15098T (95 %) and Sphingomonas adhaesiva GIFU 11458T (92 %). These strains (except Sphingopyxis terrae IFO 15098T, which was not investigated) and the novel isolate accumulated polyhydroxy- alkanoates consisting of 3-hydroxybutyric acid and 3-hydroxyvaleric acid from glucose as carbon source. The G+C content of the DNA of strain S37T was 65?5 mol%. The major cellular fatty acids of this strain were octadecenoic acid (18 : 1o7c), heptadecenoic acid (17 : 1o6c) and hexadecanoic acid (16 : 0). The results of DNA–DNA hybridization experiments and its physiological characteristics clearly distinguished the novel isolate from all known Sphingopyxis species and indicated that the strain represents a novel Sphingopyxis species. -

Estudio Del Crecimiento De Microcystis Aeruginosa Y De La Producción De Microcystina En Cultivo De Laboratorio
UNIVERSIDAD NACIONAL DE LA PLATA FACULTAD DE CIENCIAS EXACTAS DEPARTAMENTO DE CIENCIAS BIOLÓGICAS Trabajo de Tesis Doctoral ESTUDIO DEL CRECIMIENTO DE MICROCYSTIS AERUGINOSA Y DE LA PRODUCCIÓN DE MICROCYSTINA EN CULTIVO DE LABORATORIO Melina Celeste Crettaz Minaglia (Leda Giannuzzi / Darío Andrinolo) 2018 1 Dedicada a mi mamá. 2 Contenidos Resumen ........................................................................................................................................................................ 5 Introducción ................................................................................................................................................................... 9 Generalidades de las cianobacterias y cianotoxinas .................................................................................................. 9 Microcystis aeruginosa y microcistinas .................................................................................................................. 14 Factores de crecimiento de cianobacterias, en especial M. aeruginosa, y de producción de microcistinas ............ 19 Temperatura ........................................................................................................................................................ 19 Nutrientes ............................................................................................................................................................ 21 Irradiación .......................................................................................................................................................... -

Genomic Analysis of the Nitrate-Respiring Sphingopyxis Granuli (Formerly Sphingomonas Macrogoltabida) Strain TFA Inmaculada García-Romero, Antonio J
García-Romero et al. BMC Genomics (2016) 17:93 DOI 10.1186/s12864-016-2411-1 RESEARCH ARTICLE Open Access Genomic analysis of the nitrate-respiring Sphingopyxis granuli (formerly Sphingomonas macrogoltabida) strain TFA Inmaculada García-Romero, Antonio J. Pérez-Pulido, Yolanda Elisabet González-Flores, Francisca Reyes-Ramírez, Eduardo Santero and Belén Floriano* Abstract Background: Sphingomonads are Alphaproteobacteria that belong to the Sphingomonas, Novosphingobium, Sphingopyxis or Sphingobium genera, They are physiologically diverse and broadly distributed in nature, playing important roles in oligotrophic environments and in the degradation of recalcitrant polyaromatic compounds, Sphingopyxis is a poorly studied genus of which only one representative (S. alaskensis RB2256) has been deeply characterized. In this paper we analyze the genomic features of S. granuli strain TFA (formerly Sphingomonas macrogoltabida) in comparison with the available Sphingopyxis sequenced genomes, to describe common characteristics of this genus and to highlight unique characteristics of strain TFA. Results: The TFA genome has been assembled in a single circular chromosome of 4.7 Mb. Genomic sequence analysis and proteome comparison re-assigned the TFA strain to the Sphingopyxis genus and the S. granuli species. Some regions of the TFA genome show high similarity (ca. 100 %) to other bacteria and several genomic islands have been detected. Pathways for aromatic compound degradation have been predicted but no growth of TFA has been detected using these as carbon or nitrogen sources. Genes for nitrate respiration have been identified as TFA exclusive. Experimental data on anaerobic growth of TFA using nitrate as a terminal electron acceptor are also provided. Conclusions: Sphingopyxis representatives form a compact phylogenetic group (with the exception of S. -

Sphingopyxis Baekryungensis Sp. Nov., an Orange-Pigmented Bacterium Isolated from Sea Water of the Yellow Sea in Korea
International Journal of Systematic and Evolutionary Microbiology (2005), 55, 1223–1227 DOI 10.1099/ijs.0.63495-0 Sphingopyxis baekryungensis sp. nov., an orange-pigmented bacterium isolated from sea water of the Yellow Sea in Korea Jung-Hoon Yoon,1 Choong-Hwan Lee,1 Soo-Hwan Yeo2 and Tae-Kwang Oh1 Correspondence 1Laboratory of Microbial Function, Korea Research Institute of Bioscience and Biotechnology Jung-Hoon Yoon (KRIBB), PO Box 115, Yusong, Taejon, Korea [email protected] 2Center for Traditional Microorganism Resources, Keimyung University, Shindang-dong, Dalseo-gu, Daegu, Korea A Gram-negative, motile, slightly halophilic bacterial strain, SW-150T, was isolated from sea water of the Yellow Sea, Korea, and was characterized by a polyphasic taxonomic approach. Strain SW-150T grew optimally at 25–30 6C and in the presence of 2 % (w/v) NaCl. The isolate could be distinguished from other Sphingopyxis species in producing an orange pigment. It contained ubiquinone-10 as the predominant respiratory lipoquinone and C18 : 1v7c and C17 : 1v6c as the major fatty acids. No 3-hydroxy fatty acids were detected. Major polar lipids were sphingoglycolipid, diphosphatidylglycerol, phosphatidylglycerol and phosphatidylethanolamine. The DNA G+C content was 63 mol%. Comparative 16S rRNA gene sequence analyses showed that strain SW-150T was phylogenetically affiliated to the genus Sphingopyxis of the family Sphingomonadaceae. Similarity values between the 16S rRNA gene sequences of strain SW-150T and the type strains of Sphingopyxis species ranged from 91?6to94?2%, making it possible to categorize strain SW-150T as a species that is separate from previously described Sphingopyxis species. On the basis of phenotypic properties and phylogenetic distinctiveness, SW-150T (=KCTC 12231T=DSM 16222T) should be classified as the type strain of a novel Sphingopyxis species, for which the name Sphingopyxis baekryungensis sp. -

Genome-Wide Analysis Reveals Genetic Potential for Aromatic Compounds Biodegradation of Sphingopyxis
Hindawi BioMed Research International Volume 2020, Article ID 5849123, 12 pages https://doi.org/10.1155/2020/5849123 Research Article Genome-Wide Analysis Reveals Genetic Potential for Aromatic Compounds Biodegradation of Sphingopyxis Fei Yang,1 Hai Feng,1 Isaac Yaw Massey,1 Feiyu Huang,1 Jian Guo,1 and Xian Zhang 1,2 1Department of Occupational and Environmental Health, Xiangya School of Public Health, Central South University, Changsha, China 2Hunan Provincial Key Laboratory of Clinical Epidemiology, Central South University, Changsha, China Correspondence should be addressed to Xian Zhang; [email protected] Received 21 August 2019; Accepted 20 April 2020; Published 27 May 2020 Academic Editor: Kazuhisa Nishizawa Copyright © 2020 Fei Yang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Members of genus Sphingopyxis are frequently found in diverse eco-environments worldwide and have been traditionally considered to play vital roles in the degradation of aromatic compounds. Over recent decades, many aromatic-degrading Sphingopyxis strains have been isolated and recorded, but little is known about their genetic nature related to aromatic compounds biodegradation. In this study, bacterial genomes of 19 Sphingopyxis strains were used for comparative analyses. Phylogeny showed an ambiguous relatedness between bacterial strains and their habitat specificity, while clustering based on Cluster of Orthologous Groups suggested the potential link of functional profile with substrate-specific traits. Pan-genome analysis revealed that 19 individuals were predicted to share 1,066 orthologous genes, indicating a high genetic homogeneity among Sphingopyxis strains. -
Phylogenomics and Signature Proteins for the Alpha Proteobacteria and Its Main Groups Radhey S Gupta* and Amy Mok
BMC Microbiology BioMed Central Research article Open Access Phylogenomics and signature proteins for the alpha Proteobacteria and its main groups Radhey S Gupta* and Amy Mok Address: Department of Biochemistry and Biomedical Science, McMaster University, Hamilton L8N3Z5, Canada Email: Radhey S Gupta* - [email protected]; Amy Mok - [email protected] * Corresponding author Published: 28 November 2007 Received: 20 July 2007 Accepted: 28 November 2007 BMC Microbiology 2007, 7:106 doi:10.1186/1471-2180-7-106 This article is available from: http://www.biomedcentral.com/1471-2180/7/106 © 2007 Gupta and Mok; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Alpha proteobacteria are one of the largest and most extensively studied groups within bacteria. However, for these bacteria as a whole and for all of its major subgroups (viz. Rhizobiales, Rhodobacterales, Rhodospirillales, Rickettsiales, Sphingomonadales and Caulobacterales), very few or no distinctive molecular or biochemical characteristics are known. Results: We have carried out comprehensive phylogenomic analyses by means of Blastp and PSI- Blast searches on the open reading frames in the genomes of several α-proteobacteria (viz. Bradyrhizobium japonicum, Brucella suis, Caulobacter crescentus, Gluconobacter oxydans, Mesorhizobium loti, Nitrobacter winogradskyi, Novosphingobium aromaticivorans, Rhodobacter sphaeroides 2.4.1, Silicibacter sp. TM1040, Rhodospirillum rubrum and Wolbachia (Drosophila) endosymbiont). These studies have identified several proteins that are distinctive characteristics of all α-proteobacteria, as well as numerous proteins that are unique repertoires of all of its main orders (viz. -
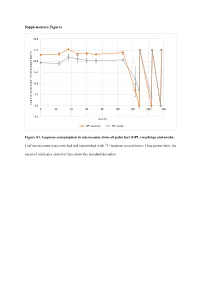
Supplementary Figures Figure S1. Isoprene Consumption In
Supplementary Figures 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppmv) 0.0 0 20406080100120140160 -5.0 Time (h) OPL washings OPL swabs Figure S1. Isoprene consumption in microcosms from oil palm leaf (OPL) washings and swabs. Leaf microcosms were enriched and replenished with 12C-isoprene several times. Data points show the mean of triplicates and error bars show the standard deviation. A 35.0 T1 T2 T3 30.0 25.0 20.0 15.0 10.0 5.0 Isoprene concentration in the headspace (ppm) headspace the in concentration Isoprene 0.0 -5.0 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 Time (h) 12C-isoprene 13C-labelled isoprene B 70.0 T1 60.0 50.0 40.0 30.0 20.0 10.0 Isoprene concentration in the headspace (ppmv) 0.0 -10.0 0 20 40 60 80 100 120 140 160 180 200 220 Time (h) 12C-isoprene 13C-labelled isoprene Figure S2. Isoprene consumption in microcosms used in DNA-SIP experiments with oil palm soil (A) and leaf washings (B). Soil (A) and leaf washings (B) microcosms were enriched and replenished with 12C- or 13C-labelled isoprene. Data points show the mean of triplicates and error bars show the standard deviation. B) Leaf washings enrichment are shown up to 220 h (T1) because of variation observed in the isoprene consumption between replicates. A 12 12 C-isoprene T1 12 C-isoprene T3 L C-isoprene T2 40 35 45 L L 35 30 40 35 30 25 30 25 20 25 20 15 20 15 15 DNA concentration(ng uL-1) DNA concentrationDNA uL-1) (ng 10 DNA concentration (ng uL-1) DNA (ng concentration 10 10 5 5 5 0 0 0 1.397 1.399 1.401 1.403 -

PHYSIOLOGICAL ADAPTATION to NUTRIENT LIMITATION in a MARINE OLIGOTROPHIC ULTRAMICROBACTERIUM Sphingopyxis Alaskensis
PHYSIOLOGICAL ADAPTATION TO NUTRIENT LIMITATION IN A MARINE OLIGOTROPHIC ULTRAMICROBACTERIUM Sphingopyxis alaskensis MARTIN OSTROWSKI A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales, Australia October 2006 UNIVERSITY OF NEW SOUTH WALES Thesis/Project Report Sheet Surname or Family name: OSTROWSKI First name: MARTIN Other name/s: - LUKE Abbreviation for degree as given in the University calendar: PhD School:BIOTECHNOLOGY AND BIOMOLECULAR SCIENCES Faculty: SCIENCE Title: PHYSIOLOGICAL ADAPTATION TO NUTRIENT LIMITATION IN A MARINE OLIGOTROPHIC ULTRAMICROBACTERIUM Sphingopyxis alaskensis Abstract 350 words maximum: Sphingopyxis (formerly Sphingomonas) alaskensis, a numerically abundant species isolated from Alaskan waters and the North Sea represents one of the only pure cultures of a typical oligotrophic ultramicrobacterium isolated from the marine environment. In this study, physiological and molecular characterization of an extinction dilution isolate from the North Pacific indicate that it is a strain of Sphingopyxis alaskenis, extending the known geographical distribution of this strain and affirming its importance as a model marine oligotroph. Given the importance of open ocean systems in climatic processes, it is clearly important to understand the physiology and underlying molecular biology of abundant species, such as S. alaskensis, and to define their role in biogeochemical processes.