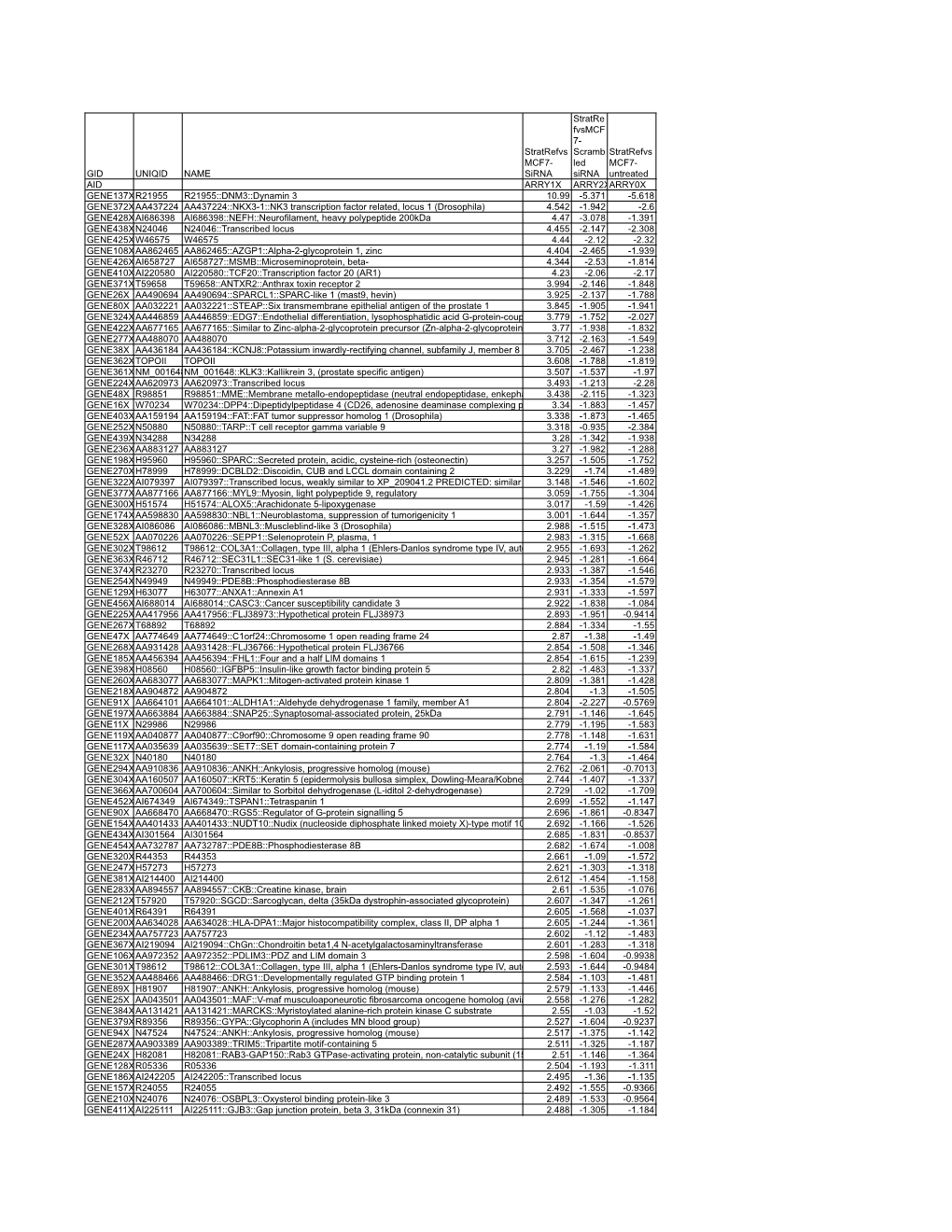
Sirna Stratre Fvsmcf 7
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis Ryan Fassnacht Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Physiology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2246 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Ryan C. Fassnacht 2010 All Rights Reserved Molecular Mechanisms Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Ryan Christopher Fassnacht, B.S. Hampden Sydney University, 2005 M.S. Virginia Commonwealth University, 2010 Director: Valeria Mas, Ph.D., Associate Professor of Surgery and Pathology Division of Transplant Department of Surgery Virginia Commonwealth University Richmond, Virginia July 9, 2010 Acknowledgement The Author wishes to thank his family and close friends for their support. He would also like to thank the members of the molecular transplant team for their help and advice. This project would not have been possible with out the help of Dr. Valeria Mas and her endearing -

1 Metabolic Dysfunction Is Restricted to the Sciatic Nerve in Experimental
Page 1 of 255 Diabetes Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy Oliver J. Freeman1,2, Richard D. Unwin2,3, Andrew W. Dowsey2,3, Paul Begley2,3, Sumia Ali1, Katherine A. Hollywood2,3, Nitin Rustogi2,3, Rasmus S. Petersen1, Warwick B. Dunn2,3†, Garth J.S. Cooper2,3,4,5* & Natalie J. Gardiner1* 1 Faculty of Life Sciences, University of Manchester, UK 2 Centre for Advanced Discovery and Experimental Therapeutics (CADET), Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre, Manchester, UK 3 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, UK 4 School of Biological Sciences, University of Auckland, New Zealand 5 Department of Pharmacology, Medical Sciences Division, University of Oxford, UK † Present address: School of Biosciences, University of Birmingham, UK *Joint corresponding authors: Natalie J. Gardiner and Garth J.S. Cooper Email: [email protected]; [email protected] Address: University of Manchester, AV Hill Building, Oxford Road, Manchester, M13 9PT, United Kingdom Telephone: +44 161 275 5768; +44 161 701 0240 Word count: 4,490 Number of tables: 1, Number of figures: 6 Running title: Metabolic dysfunction in diabetic neuropathy 1 Diabetes Publish Ahead of Print, published online October 15, 2015 Diabetes Page 2 of 255 Abstract High glucose levels in the peripheral nervous system (PNS) have been implicated in the pathogenesis of diabetic neuropathy (DN). However our understanding of the molecular mechanisms which cause the marked distal pathology is incomplete. Here we performed a comprehensive, system-wide analysis of the PNS of a rodent model of DN. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Microrna Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton
cells Review MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton , , Karen Uray * y , Evelin Major and Beata Lontay * y Department of Medical Chemistry, Faculty of Medicine, University of Debrecen, 4032 Debrecen, Hungary; [email protected] * Correspondence: [email protected] (K.U.); [email protected] (B.L.); Tel.: +36-52-412345 (K.U. & B.L.) The authors contributed equally to the manuscript. y Received: 11 June 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: MicroRNAs (miRNAs) are key modulators of post-transcriptional gene regulation in a plethora of processes, including actin–myosin cytoskeleton dynamics. Recent evidence points to the widespread effects of miRNAs on actin–myosin cytoskeleton dynamics, either directly on the expression of actin and myosin genes or indirectly on the diverse signaling cascades modulating cytoskeletal arrangement. Furthermore, studies from various human models indicate that miRNAs contribute to the development of various human disorders. The potentially huge impact of miRNA-based mechanisms on cytoskeletal elements is just starting to be recognized. In this review, we summarize recent knowledge about the importance of microRNA modulation of the actin–myosin cytoskeleton affecting physiological processes, including cardiovascular function, hematopoiesis, podocyte physiology, and osteogenesis. Keywords: miRNA; actin; myosin; actin–myosin complex; Rho kinase; cancer; smooth muscle; hematopoiesis; stress fiber; gene expression; cardiovascular system; striated muscle; muscle cell differentiation; therapy 1. Introduction Actin–myosin interactions are the primary source of force generation in mammalian cells. Actin forms a cytoskeletal network and the myosin motor proteins pull actin filaments to produce contractile force. All eukaryotic cells contain an actin–myosin network inferring contractile properties to these cells. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Non-Muscle Myosin 2A (NM2A): Structure, Regulation and Function
cells Review Non-Muscle Myosin 2A (NM2A): Structure, Regulation and Function Cláudia Brito 1,2 and Sandra Sousa 1,* 1 Group of Cell Biology of Bacterial Infections, i3S-Instituto de Investigação e Inovação em Saúde, IBMC, Universidade do Porto, 4200-135 Porto, Portugal; [email protected] 2 Programa Doutoral em Biologia Molecular e Celular (MCBiology), Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, 4099-002 Porto, Portugal * Correspondence: [email protected] Received: 19 May 2020; Accepted: 29 June 2020; Published: 1 July 2020 Abstract: Non-muscle myosin 2A (NM2A) is a motor cytoskeletal enzyme with crucial importance from the early stages of development until adulthood. Due to its capacity to convert chemical energy into force, NM2A powers the contraction of the actomyosin cytoskeleton, required for proper cell division, adhesion and migration, among other cellular functions. Although NM2A has been extensively studied, new findings revealed that a lot remains to be discovered concerning its spatiotemporal regulation in the intracellular environment. In recent years, new functions were attributed to NM2A and its activity was associated to a plethora of illnesses, including neurological disorders and infectious diseases. Here, we provide a concise overview on the current knowledge regarding the structure, the function and the regulation of NM2A. In addition, we recapitulate NM2A-associated diseases and discuss its potential as a therapeutic target. Keywords: non-muscle myosin 2A (NM2A); NM2A activity regulation; NM2A filament assembly; actomyosin cytoskeleton; cell migration; cell adhesion; plasma membrane blebbing 1. Superfamily of Myosins The cell cytoskeleton is an interconnected and dynamic network of filaments essential for intracellular organization and cell shape maintenance. -

Homozygous Deletion in MYL9 Expands the Molecular Basis of Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome
European Journal of Human Genetics (2018) 26:669–675 https://doi.org/10.1038/s41431-017-0055-5 ARTICLE Homozygous deletion in MYL9 expands the molecular basis of megacystis–microcolon–intestinal hypoperistalsis syndrome 1 2 3 3 2 1 Carolina Araujo Moreno ● Nara Sobreira ● Elizabeth Pugh ● Peng Zhang ● Gary Steel ● Fábio Rossi Torres ● Denise Pontes Cavalcanti1 Received: 16 June 2017 / Revised: 14 November 2017 / Accepted: 18 November 2017 / Published online: 16 February 2018 © European Society of Human Genetics 2018 Abstract Megacystis–microcolon–intestinal hypoperistalsis syndrome (MMIHS) is a severe disease characterized by functional obstruction in the urinary and gastrointestinal tract. The molecular basis of this condition started to be defined recently, and the genes related to the syndrome (ACTG2—heterozygous variant in sporadic cases; and MYH11 (myosin heavy chain 11), LMOD1 (leiomodin 1) and MYLK (myosin light chain (MLC) kinase)—autosomal recessive inheritance), encode proteins involved in the smooth muscle contraction, supporting a myopathic basis for the disease. In the present article, we described a family with two affected siblings with MMIHS born to consanguineous parents and the molecular investigation performed fi 1234567890 to de ne the genetic etiology. Previous whole exome sequencing of the affected child and parents did not identify a candidate gene for the disease in this family, but now we present a reanalysis of the data that led to the identification of a homozygous deletion encompassing the last exon of MYL9 (myosin regulatory light chain 9) in the affected individual. MYL9 gene encodes a regulatory myosin MLC and the phosphorylation of this protein is a crucial step in the contraction process of smooth muscle cell. -

Identification of the Fatty Acid Synthase Interaction Network Via Itraq-Based Proteomics Indicates the Potential Molecular Mecha
Huang et al. Cancer Cell Int (2020) 20:332 https://doi.org/10.1186/s12935-020-01409-2 Cancer Cell International PRIMARY RESEARCH Open Access Identifcation of the fatty acid synthase interaction network via iTRAQ-based proteomics indicates the potential molecular mechanisms of liver cancer metastasis Juan Huang1, Yao Tang1, Xiaoqin Zou1, Yi Lu1, Sha She1, Wenyue Zhang1, Hong Ren1, Yixuan Yang1,2* and Huaidong Hu1,2* Abstract Background: Fatty acid synthase (FASN) is highly expressed in various types of cancer and has an important role in carcinogenesis and metastasis. To clarify the mechanisms of FASN in liver cancer invasion and metastasis, the FASN protein interaction network in liver cancer was identifed by targeted proteomic analysis. Methods: Wound healing and Transwell assays was performed to observe the efect of FASN during migration and invasion in liver cancer. Isobaric tags for relative and absolute quantitation (iTRAQ)-based mass spectrometry were used to identify proteins interacting with FASN in HepG2 cells. Diferential expressed proteins were validated by co-immunoprecipitation, western blot analyses and confocal microscopy. Western blot and reverse transcription- quantitative polymerase chain reaction (RT-qPCR) were performed to demonstrate the mechanism of FASN regulating metastasis. Results: FASN knockdown inhibited migration and invasion of HepG2 and SMMC7721 cells. A total of, 79 proteins interacting with FASN were identifed. Additionally, gene ontology term enrichment analysis indicated that the majority of biological regulation and cellular processes that the FASN-interacting proteins were associated with. Co- precipitation and co-localization of FASN with fascin actin-bundling protein 1 (FSCN1), signal-induced proliferation- associated 1 (SIPA1), spectrin β, non-erythrocytic 1 (SPTBN1) and CD59 were evaluated. -

Bioinformatic Analysis Reveals the Importance of Epithelial-Mesenchymal Transition in the Development of Endometriosis
www.nature.com/scientificreports OPEN Bioinformatic analysis reveals the importance of epithelial- mesenchymal transition in the development of endometriosis Meihong Chen1,6, Yilu Zhou2,3,6, Hong Xu4, Charlotte Hill2, Rob M. Ewing2,3, Deming He1, Xiaoling Zhang1 ✉ & Yihua Wang2,3,5 ✉ Background: Endometriosis is a frequently occurring disease in women, which seriously afects their quality of life. However, its etiology and pathogenesis are still unclear. Methods: To identify key genes/ pathways involved in the pathogenesis of endometriosis, we recruited 3 raw microarray datasets (GSE11691, GSE7305, and GSE12768) from Gene Expression Omnibus database (GEO), which contain endometriosis tissues and normal endometrial tissues. We then performed in-depth bioinformatic analysis to determine diferentially expressed genes (DEGs), followed by gene ontology (GO), Hallmark pathway enrichment and protein-protein interaction (PPI) network analysis. The fndings were further validated by immunohistochemistry (IHC) staining in endometrial tissues from endometriosis or control patients. Results: We identifed 186 DEGs, of which 118 were up-regulated and 68 were down-regulated. The most enriched DEGs in GO functional analysis were mainly associated with cell adhesion, infammatory response, and extracellular exosome. We found that epithelial-mesenchymal transition (EMT) ranked frst in the Hallmark pathway enrichment. EMT may potentially be induced by infammatory cytokines such as CXCL12. IHC confrmed the down-regulation of E-cadherin (CDH1) and up-regulation of CXCL12 in endometriosis tissues. Conclusions: Utilizing bioinformatics and patient samples, we provide evidence of EMT in endometriosis. Elucidating the role of EMT will improve the understanding of the molecular mechanisms involved in the development of endometriosis. Endometriosis is a frequently occurring gynaecological disease characterised by chronic pelvic pain, dysmenor- rhea and infertility1. -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function.