Dissertation
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Genome of Prasinoderma Coloniale Unveils the Existence of a Third Phylum Within Green Plants
SUPPLEMENTARY INFORMATIONARTICLES https://doi.org/10.1038/s41559-020-1221-7 In the format provided by the authors and unedited. The genome of Prasinoderma coloniale unveils the existence of a third phylum within green plants Linzhou Li1,2,13, Sibo Wang1,3,13, Hongli Wang1,4, Sunil Kumar Sahu 1, Birger Marin 5, Haoyuan Li1, Yan Xu1,4, Hongping Liang1,4, Zhen Li 6, Shifeng Cheng1, Tanja Reder5, Zehra Çebi5, Sebastian Wittek5, Morten Petersen3, Barbara Melkonian5,7, Hongli Du8, Huanming Yang1, Jian Wang1, Gane Ka-Shu Wong 1,9, Xun Xu 1,10, Xin Liu 1, Yves Van de Peer 6,11,12 ✉ , Michael Melkonian5,7 ✉ and Huan Liu 1,3 ✉ 1State Key Laboratory of Agricultural Genomics, BGI-Shenzhen, Shenzhen, China. 2Department of Biotechnology and Biomedicine, Technical University of Denmark, Lyngby, Denmark. 3Department of Biology, University of Copenhagen, Copenhagen, Denmark. 4BGI Education Center, University of Chinese Academy of Sciences, Shenzhen, China. 5Institute for Plant Sciences, Department of Biological Sciences, University of Cologne, Cologne, Germany. 6Department of Plant Biotechnology and Bioinformatics (Ghent University) and Center for Plant Systems Biology, Ghent, Belgium. 7Central Collection of Algal Cultures, Faculty of Biology, University of Duisburg-Essen, Essen, Germany. 8School of Biology and Biological Engineering, South China University of Technology, Guangzhou, China. 9Department of Biological Sciences and Department of Medicine, University of Alberta, Edmonton, Alberta, Canada. 10Guangdong Provincial Key Laboratory of Genome Read and Write, BGI-Shenzhen, Shenzhen, China. 11College of Horticulture, Nanjing Agricultural University, Nanjing, China. 12Centre for Microbial Ecology and Genomics, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa. -

Phylogenetic and Evolutionary Patterns in Microbial Carotenoid Biosynthesis Are Revealed by Comparative Genomics
Phylogenetic and Evolutionary Patterns in Microbial Carotenoid Biosynthesis Are Revealed by Comparative Genomics Jonathan L. Klassen* Department of Biological Sciences, University of Alberta, Edmonton, Alberta, Canada Abstract Background: Carotenoids are multifunctional, taxonomically widespread and biotechnologically important pigments. Their biosynthesis serves as a model system for understanding the evolution of secondary metabolism. Microbial carotenoid diversity and evolution has hitherto been analyzed primarily from structural and biosynthetic perspectives, with the few phylogenetic analyses of microbial carotenoid biosynthetic proteins using either used limited datasets or lacking methodological rigor. Given the recent accumulation of microbial genome sequences, a reappraisal of microbial carotenoid biosynthetic diversity and evolution from the perspective of comparative genomics is warranted to validate and complement models of microbial carotenoid diversity and evolution based upon structural and biosynthetic data. Methodology/Principal Findings: Comparative genomics were used to identify and analyze in silico microbial carotenoid biosynthetic pathways. Four major phylogenetic lineages of carotenoid biosynthesis are suggested composed of: (i) Proteobacteria; (ii) Firmicutes; (iii) Chlorobi, Cyanobacteria and photosynthetic eukaryotes; and (iv) Archaea, Bacteroidetes and two separate sub-lineages of Actinobacteria. Using this phylogenetic framework, specific evolutionary mechanisms are proposed for carotenoid desaturase CrtI-family -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

P^I"~ SUBMITTED to the DEPARTMENT of L \IL.~D EARTH, ATMOSPHERIC and PLANETARY SCIENCES in PARTIAL FULFILLMENT of the REQUIREMENTS for the DEGREE OF
Molecular studies of the sources and significance of archaeal lipids in the oceans by Sara Ann Lincoln MIA IsJ:S' iNSTITUTE B.S. Geosciences B.S. Geological Oceanography L T University of Rhode Island (2006) P^I"~ SUBMITTED TO THE DEPARTMENT OF L \IL.~d EARTH, ATMOSPHERIC AND PLANETARY SCIENCES IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN GEOCHEMISTRY SEPTEMBER 2013 © Massachusetts Institute of Technology All rights reserved. Author:................. Department of Earth, Atmospheric and Planetary Sciences Certified by:........................... Roger E. Summons Professor of Geobiology Department of Earth, Atmospheric and Planetary Sciences Thesis supervisor ..................#......r....... ........................................................ Edward F. DeLong Morton and Claire Goulder amily P fessor in Environmental Systems Depar e Civil and Environmental Engineering 7) Thesis supervisor Accepted by: ...................................... Robert van der Hilst Schlumberger Professor of Earth Sciences Head, Department of Earth, Atmospheric and Planetary Sciences THIS PAGE INTENTIONALLY LEFT BLANK Molecular studies of the sources and significance of archaeal lipids in the oceans by Sara Ann Lincoln Submitted to the Department of Earth, Atmospheric and Planetary Sciences on July 29, 2013 in partial fulfillment of the requirements for the Degree of Doctor of Philosophy in Geochemistry ABSTRACT Marine archaea are ubiquitous and abundant in the modem oceans and have a geologic record extending >100 million years. However, factors influencing the populations of the major clades - chemolithoautotrophic Marine Group I Thaumarchaeota (MG-I) and heterotrophic Marine Group II Euryarchaeota (MG-II) - and their membrane lipid signatures are not well understood. Here, I paired techniques of organic geochemistry and molecular biology to explore the sources and significance of archaeal tetraether lipids in the marine water column. -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

Biogeography and Habitat Modelling of High-Alpine Bacteria
ARTICLE Received 7 Apr 2010 | Accepted 15 Jul 2010 | Published 10 Aug 2010 DOI: 10.1038/ncomms1055 Biogeography and habitat modelling of high-alpine bacteria Andrew J. King1 , Kristen R. Freeman1 , Katherine F. McCormick1 , Ryan C. Lynch1 , Catherine Lozupone2 , 3 , Rob Knight 2 & Steven K. Schmidt1 Soil microorganisms dominate terrestrial biogeochemical cycles; however, we know very little about their spatial distribution and how changes in the distributions of specifi c groups of microbes translate into landscape and global patterns of biogeochemical processes. In this paper, we use a nested sampling scheme at scales ranging from 2 to 2,000 m to show that bacteria have signifi cant spatial autocorrelation in community composition up to a distance of 240 m, and that this pattern is driven by changes in the relative abundance of specifi c bacterial clades across the landscape. Analysis of clade habitat distribution models and spatial co-correlation maps identifi ed soil pH, plant abundance and snow depth as major variables structuring bacterial communities across this landscape, and revealed an unexpected and important oligotrophic niche for the Rhodospirillales in soil. Furthermore, our global analysis of high-elevation soils from the Andes, Rockies, Himalayas and Alaskan range shows that habitat distribution models for bacteria have a strong predictive power across the entire globe. 1 Department of Ecology and Evolutionary Biology, University of Colorado , Boulder , Colorado 80309 , USA . 2 Department of Chemistry and Biochemistry, University of Colorado , Boulder , Colorado 80309 , USA . 3 Washington University School of Medicine , St Louis , Missouri 63108 , USA . Correspondence and requests for materials should be addressed to S.K.S. -

Genetic and Functional Studies of the Mip Protein of Legionella
t1.ì. Genetic and Functional Studies of the Mip Protein of Legionella Rodney Mark Ratcliff, BSc (Hons)' MASM Infectious Diseases Laboratories Institute of Medical and Veterinary Science and Department of Microbiology and Immunology UniversitY of Adelaide. Adelaide, South Australia A thesis submitted to the University of Adelaide for the degree of I)octor of Philosophy 15'h March 2000 amended 14th June 2000 Colonies of several Legionella strains on charcoal yeast extract agar (CYE) after 4 days incubation at 37"C in air. Various magnifications show typical ground-glass opalescent appearance. Some pure strains exhibit pleomorphic growth or colour. The top two photographs demonstrate typical red (LH) and blue-white (RH) fluorescence exhibited by some species when illuminated by a Woods (IJV) Lamp. * t Table of Contents .1 Chapter One: Introduction .1 Background .............'. .2 Morphology and TaxonomY J Legionellosis ............. 5 Mode of transmission "..'....'. 7 Environmental habitat 8 Interactions between Legionella and phagocytic hosts 9 Attachment 11 Engulfment and internalisation.'.. 13 Intracellular processing 13 Intracellular replication of legionellae .. " "' " "' 15 Host cell death and bacterial release 18 Virulence (the Genetic factors involved with intracellular multiplication and host cell killing .20 icm/dot system) Legiolysin .25 Msp (Znn* metaloprotease) ...'..... .25 .28 Lipopolysaccharide .29 The association of flagella with disease.. .30 Type IV fimbriae.... .31 Major outer membrane proteins....'.......'. JJ Heat shock proteins'.'. .34 Macrophage infectivity potentiator (Mip) protein Virulenceiraits of Legionella species other than L. pneumophila..........' .39 phylogeny .41 Chapter One (continued): Introduction to bacterial classification and .41 Identificati on of Legionella...'.,..'.. .46 Phylogeny .52 Methods of phylogenetic analysis' .53 Parsimony methods.'.. .55 Distance methods UPGMA cluster analYsis.'.'... -

Electronic Laboratory Reporting Use Case January 2011
ILLINOIS HEALTH INFORMATION EXCHANGE Electronic Laboratory Reporting Use Case Electronic Laboratory Reporting and Health Information Exchange Illinois Health Information Exchange Public Health Work Group January 2011 Electronic Laboratory Reporting Use Case January 2011 Table of Contents 1.0 Executive Summary……………………………………………………………………….3 2.0 Introduction…………………………………………………………………...……………..5 3.0 Scope……………………………………..………………………………………………………5 4.0 Use Case Stakeholders…………………………………………………………….….....6 5.0 Issues and Obstacles……………………………………………………………………...8 6.0 Use Case Pre-Conditions .………………….…………………………………………...8 7.0 Use Case Post-Conditions.……………………………………………………………...9 8.0 Detailed Scenarios/Technical Specifications.………………………………10 9.0 Validation and Certification………………………………………………………...12 Appendix ………………………………………………………………………………………….....13 Page 2 Electronic Laboratory Reporting Use Case January 2011 1.0 Executive Summary This Use Case is a product of the Public Health Work Group (PHWG) of the Illinois Health Information Exchange (HIE) Advisory Committee. The Illinois HIE Advisory Committee was constituted as the diverse public healthcare stakeholder body providing input and recommendations on the creation of the Illinois Health Information Exchange Authority (“the Authority”) as the Illinois vehicle for designing and implementing electronic health information exchange in Illinois. The establishment of the Authority marks the formal transition of the work of the HIE Advisory Committee and the Work Groups into alignment with the provisions of Illinois -

WO 2016/188962 Al 1 December 2016 (01.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/188962 Al 1 December 2016 (01.12.2016) P O P C T (51) International Patent Classification: (74) Agents: GOODFELLOW, Hugh Robin et al; Carpmaels C12Q 1/68 (2006.01) & Ransford LLP, One Southampton Row, London WC1B 5HA (GB). (21) International Application Number: PCT/EP2016/061599 (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (22) Date: International Filing AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 23 May 20 16 (23.05.2016) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (25) Filing Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (26) Publication Language: English KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (30) Priority Data: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 1508860.2 22 May 2015 (22.05.2015) GB PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (71) Applicant: NATIONAL UNIVERSITY OF IRELAND, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. GALWAY [IE/IE]; University Road, Galway (IE). (84) Designated States (unless otherwise indicated, for every (72) Inventors: REDDINGTON, Kate Mary; Deerpack East, kind of regional protection available): ARIPO (BW, GH, Newport Road, Westport, Co. -

EVALUATION of the P45 MOBILE INTEGRATIVE ELEMENT and ITS ROLE IN
EVALUATION OF THE p45 MOBILE INTEGRATIVE ELEMENT AND ITS ROLE IN Legionella pneumophila VIRULENCE A Dissertation by LANETTE M. CHRISTENSEN Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Jeffrey D. Cirillo Committee Members, James Samuel Jon Skare Farida Sohrabji Head of Program, Warren Zimmer May 2018 Major Subject: Medical Sciences Copyright 2018 Lanette Christensen ABSTRACT Legionella pneumophila are aqueous environmental bacilli that live within protozoal species and cause a potentially fatal form of pneumonia called Legionnaires’ disease. Not all L. pneumophila strains have the same capacity to cause disease in humans. The majority of strains that cause clinically relevant Legionnaires’ disease harbor the p45 mobile integrative genomic element. Contribution of the p45 element to L. pneumophila virulence and ability to withstand environmental stress were addressed in this study. The L. pneumophila Philadelphia-1 (Phil-1) mobile integrative element, p45, was transferred into the attenuated strain Lp01 via conjugation, designating p45 an integrative conjugative element (ICE). The resulting trans-conjugate, Lp01+p45, was compared with strains Phil-1 and Lp01 to assess p45 in virulence using a guinea pig model infected via aerosol. The p45 element partially recovered the loss of virulence in Lp01 compared to that of Phil-1 evident in morbidity, mortality, and bacterial burden in the lungs at the time of death. This phenotype was accompanied by enhanced expression of type II interferon in the lungs and spleens 48 hours after infection, independent of bacterial burden. -
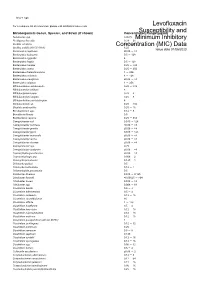
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

Aquascreen® Legionella Species Qpcr Detection Kit
AquaScreen® Legionella species qPCR Detection Kit INSTRUCTIONS FOR USE FOR USE IN RESEARCH AND QUALITY CONTROL Symbols Lot No. Cat. No. Expiry date Storage temperature Number of reactions Manufacturer INDICATION The AquaScreen® Legionella species qPCR Detection kit is specifically designed for the quantitative detection of several Legionella species in water samples prepared with the AquaScreen® FastExt- ract kit. Its design complies with the requirements of AFNOR T90-471 and ISO/TS 12869:2012. Legionella are ubiquitous bacteria in surface water and moist soil, where they parasitize protozoa. The optimal growth temperature lies between +15 and +45 °C, whereas these gram-negative bacteria are dormant below 20 °C and do not survive above 60 °C. Importantly, Legionella are well-known as opportunistic intracellular human pathogens causing Legionnaires’ disease and Pontiac fever. The transmission occurs through inhalation of contami- nated aerosols generated by an infected source (e.g. human-made water systems like shower- heads, sink faucets, heaters, cooling towers, and many more). In order to efficiently prevent Legionella outbreaks, water safety control measures need syste- matic application but also reliable validation by fast Legionella testing. TEST PRINCIPLE The AquaScreen® Legionella species Kit uses qPCR for quantitative detection of legionella in wa- ter samples. In contrast to more time-consuming culture-based methods, AquaScreen® assays need less than six hours including sample preparation and qPCR to reliably detect Legionella. Moreover, the AquaScreen® qPCR assay has proven excellent performance in terms of specificity and sensitivity: other bacterial genera remain undetected whereas linear quantification is obtai- ned up to 1 x 106 particles per sample, therefore requiring no material dilution.