Upper/Lower Respiratory Tract Microbiome Changes in AECOPD and COPD
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Identification of Pasteurella Species and Morphologically Similar Organisms
UK Standards for Microbiology Investigations Identification of Pasteurella species and Morphologically Similar Organisms Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 1 of 28 © Crown copyright 2015 Identification of Pasteurella species and Morphologically Similar Organisms Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 2 of 28 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Pasteurella species and Morphologically Similar Organisms Contents ACKNOWLEDGMENTS ......................................................................................................... -

Microbial Biofilms – Veronica Lazar and Eugenia Bezirtzoglou
MEDICAL SCIENCES – Microbial Biofilms – Veronica Lazar and Eugenia Bezirtzoglou MICROBIAL BIOFILMS Veronica Lazar University of Bucharest, Faculty of Biology, Dept. of Microbiology, 060101 Aleea Portocalelor No. 1-3, Sector 6, Bucharest, Romania; Eugenia Bezirtzoglou Democritus University of Thrace - Faculty of Agricultural Development, Dept. of Microbiology, Orestiada, Greece Keywords: Microbial adherence to cellular/inert substrata, Biofilms, Intercellular communication, Quorum Sensing (QS) mechanism, Dental plaque, Tolerance to antimicrobials, Anti-biofilm strategies, Ecological and biotechnological significance of biofilms Contents 1. Introduction 2. Definition 3. Microbial Adherence 4. Development, Architecture of a Mature Biofilm and Properties 5. Intercellular Communication: Intra-, Interspecific and Interkingdom Signaling, By QS Mechanism and Implications 6. Medical Significance of Microbial Biofilms Formed on Cellular Substrata and Medical Devices 6.1. Microbial Biofilms on Medical Devices 6.2. Microorganisms - Biomaterial Interactions 6.3. Phenotypical Resistance or Tolerance to Antimicrobials; Mechanisms of Tolerance 7. New Strategies for Prevention and Treatment of Biofilm Associated Infections 8. Ecological Significance 9. Biotechnological / Industrial Applications 10. Conclusion Acknowledgments Glossary Bibliography Biographical Sketches UNESCO – EOLSS Summary A biofilm is a sessileSAMPLE microbial community coCHAPTERSmposed of cells embedded in a matrix of exopolysaccharide matrix attached to a substratum or interface. Biofilms -

Yopb and Yopd Constitute a Novel Class of Yersinia Yop Proteins
INFECTION AND IMMUNITY, Jan. 1993, p. 71-80 Vol. 61, No. 1 0019-9567/93/010071-10$02.00/0 Copyright © 1993, American Society for Microbiology YopB and YopD Constitute a Novel Class of Yersinia Yop Proteins SEBASTIAN HAKANSSON,1 THOMAS BERGMAN,1 JEAN-CLAUDE VANOOTEGHEM, 2 GUY CORNELIS,2 AND HANS WOLF-WATZ1* Department of Cell and Molecular Biology, University of Umed, S-901 87 Umed, Sweden,' and Microbial Pathogenesis Unit, Intemnational Institute of Cellular and Molecular Pathology and Faculte6 de Medecine, Universite Catholique de Louvain, B-1200 Brussels, Belgium2 Received 21 May 1992/Accepted 21 October 1992 Virulent Yersinia species harbor a common plasmid that encodes essential virulence determinants (Yersinia outer proteins [Yops]), which are regulated by the extracellular stimuli Ca2" and temperature. The V-antigen-encoding operon has been shown to be involved in the Ca2 -regulated negative pathway. The genetic organization of the V-antigen operon and the sequence of the krGVH genes were recently presented. The V-antigen operon was shown to be a polycistronic operon having the gene order kcrGVH-yopBD (T. Bergman, S. Hakansson, A. Forsberg, L. Norlander, A. Maceliaro, A. Backman, I. Bolin, and H. Wolf-Watz, J. Bacteriol. 173:1607-1616, 1991; S. B. Price, K. Y. Leung, S. S. Barve, and S. C. Straley, J. Bacteriol. 171:5646-5653, 1989). We present here the sequence of the distal part of the V-antigen operons of Yersinia pseudotuberculosis and Yersinia enterocolitica. The sequence information encompasses theyopB andyopD genes and a downstream region in both species. We conclude that the V-antigen operon ends with theyopD gene. -

Wedding Higher Taxonomic Ranks with Metabolic Signatures Coded in Prokaryotic Genomes
Wedding higher taxonomic ranks with metabolic signatures coded in prokaryotic genomes Gregorio Iraola*, Hugo Naya* Corresponding authors: E-mail: [email protected], [email protected] This PDF file includes: Supplementary Table 1 Supplementary Figures 1 to 4 Supplementary Methods SUPPLEMENTARY TABLES Supplementary Tab. 1 Supplementary Tab. 1. Full prediction for the set of 108 external genomes used as test. genome domain phylum class order family genus prediction alphaproteobacterium_LFTY0 Bacteria Proteobacteria Alphaproteobacteria Rhodobacterales Rhodobacteraceae Unknown candidatus_nasuia_deltocephalinicola_PUNC_CP013211 Bacteria Proteobacteria Gammaproteobacteria Unknown Unknown Unknown candidatus_sulcia_muelleri_PUNC_CP013212 Bacteria Bacteroidetes Flavobacteriia Flavobacteriales NA Candidatus Sulcia deinococcus_grandis_ATCC43672_BCMS0 Bacteria Deinococcus-Thermus Deinococci Deinococcales Deinococcaceae Deinococcus devosia_sp_H5989_CP011300 Bacteria Proteobacteria Unknown Unknown Unknown Unknown micromonospora_RV43_LEKG0 Bacteria Actinobacteria Actinobacteria Micromonosporales Micromonosporaceae Micromonospora nitrosomonas_communis_Nm2_CP011451 Bacteria Proteobacteria Betaproteobacteria Nitrosomonadales Nitrosomonadaceae Unknown nocardia_seriolae_U1_BBYQ0 Bacteria Actinobacteria Actinobacteria Corynebacteriales Nocardiaceae Nocardia nocardiopsis_RV163_LEKI01 Bacteria Actinobacteria Actinobacteria Streptosporangiales Nocardiopsaceae Nocardiopsis oscillatoriales_cyanobacterium_MTP1_LNAA0 Bacteria Cyanobacteria NA Oscillatoriales -

International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/Ijsem.0.001485
International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/ijsem.0.001485 Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Mobolaji Adeolu,† Seema Alnajar,† Sohail Naushad and Radhey S. Gupta Correspondence Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Radhey S. Gupta L8N 3Z5, Canada [email protected] Understanding of the phylogeny and interrelationships of the genera within the order ‘Enterobacteriales’ has proven difficult using the 16S rRNA gene and other single-gene or limited multi-gene approaches. In this work, we have completed comprehensive comparative genomic analyses of the members of the order ‘Enterobacteriales’ which includes phylogenetic reconstructions based on 1548 core proteins, 53 ribosomal proteins and four multilocus sequence analysis proteins, as well as examining the overall genome similarity amongst the members of this order. The results of these analyses all support the existence of seven distinct monophyletic groups of genera within the order ‘Enterobacteriales’. In parallel, our analyses of protein sequences from the ‘Enterobacteriales’ genomes have identified numerous molecular characteristics in the forms of conserved signature insertions/deletions, which are specifically shared by the members of the identified clades and independently support their monophyly and distinctness. Many of these groupings, either in part or in whole, have been recognized in previous evolutionary studies, but have not been consistently resolved as monophyletic entities in 16S rRNA gene trees. The work presented here represents the first comprehensive, genome- scale taxonomic analysis of the entirety of the order ‘Enterobacteriales’. -

Pan-Genome Analyses Identify Lineage
University of Rhode Island DigitalCommons@URI Cell and Molecular Biology Faculty Publications Cell and Molecular Biology 2014 Pan-Genome Analyses Identify Lineage- and Niche-Specific aM rkers of Evolution and Adaptation in Epsilonproteobacteria Ying Zhang University of Rhode Island, [email protected] Stefan M. Sievert Creative Commons License Creative Commons License This work is licensed under a Creative Commons Attribution 3.0 License. Follow this and additional works at: https://digitalcommons.uri.edu/cmb_facpubs Citation/Publisher Attribution Zhang Y., Sievert S.M. (2014). "Pan-genome analyses identify lineage- and niche-specific am rkers of evolution and adaptation in Epsilonproteobacteria." Frontiers in Microbiology. 5: 110. Available at: http://dx.doi.org/10.3389/fmicb.2014.00110 This Article is brought to you for free and open access by the Cell and Molecular Biology at DigitalCommons@URI. It has been accepted for inclusion in Cell and Molecular Biology Faculty Publications by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. ORIGINAL RESEARCH ARTICLE published: 19 March 2014 MICROBIOLOGY doi: 10.3389/fmicb.2014.00110 Pan-genome analyses identify lineage- and niche-specific markers of evolution and adaptation in Epsilonproteobacteria Ying Zhang*† and Stefan M. Sievert Biology Department, Woods Hole Oceanographic Institution, Woods Hole, MA, USA Edited by: The rapidly increasing availability of complete bacterial genomes has created new Martin G. Klotz, University of North opportunities for reconstructing bacterial evolution, but it has also highlighted the difficulty Carolina at Charlotte, USA to fully understand the genomic and functional variations occurring among different Reviewed by: lineages. Using the class Epsilonproteobacteria as a case study, we investigated the Barbara J. -

Yersinia Enterocolitica Monographic Study
Tirziu E. et. al./Scientific Papers: Animal Science and Biotechnologies, 2011, 44 (2) Yersinia enterocolitica Monographic Study Emil Tirziu, Ciceronis Cumpanasoiu, Radu Valentin Gros, Monica Seres Faculty of Veterinary Medicine, 300645, Timisoara, Calea Aradului, 119, Romania Abstract Germs from Yersinia genus have a vast ecologic niche, being met at different domestic and wild animal species, but also in food, water and soil. The majority of yersinis live in the digestive tract of human and numerous animal species, especially rodents, but also in soil, plant debris, waters etc. Numerous species of Yersinia genus could produce characteristic infections in human, the main source of infections is represented by rodents and hematophagous insects or, more frequently, by water or contaminated food. In a 1999 study, Mead and coauthors established that the Yersinia enterocolitica prevalence in food, in USA, is around 90%. Foods of animal origin more frequently contaminated with Yersinia enterocolitica are: pork, poultry, beef and lamb meat, milk, ice-cream, sea fruits etc., among them pork meat and milk represents the sources of the most numerous toxi-infection outbreaks in human, in different world regions. Bacteria determine infections which interest the digestive tract in numerous animal species and human, with diarrhea, lymphadenitis, pneumonia and abortion are the most important symptoms. Yersinia enterocolitica enter the human body regularly by oral ingestion, and localize itself with predilection in the distal portion of the ileum and at the ileocaecal appendix and proximal colon level, were determine a terminal ileitis with lymphadenitis, acute enterocolitis, and secondary accompanied with nodosum erythema, poliartritis that could be complicated with septicemia, sometimes leading to death. -

Intrahepatic Bacterial Metataxonomic Signature in Non-Alcoholic Fatty Liver
Hepatology ORIGINAL RESEARCH Intrahepatic bacterial metataxonomic signature in Gut: first published as 10.1136/gutjnl-2019-318811 on 2 January 2020. Downloaded from non- alcoholic fatty liver disease Silvia Sookoian ,1,2 Adrian Salatino,1,3 Gustavo Osvaldo Castaño,4 Maria Silvia Landa,1,3 Cinthia Fijalkowky,1,3 Martin Garaycoechea,5 Carlos Jose Pirola 1,3 ► Additional material is ABSTRact published online only. To view Objective We aimed to characterise the liver tissue Significance of this study please visit the journal online bacterial metataxonomic signature in two independent (http:// dx. doi. org/ 10. 1136/ What is already known on this subject? gutjnl- 2019- 318811). cohorts of patients with biopsy- proven non- alcoholic fatty liver disease (NAFLD) diagnosis, as differences in ► The natural history of non- alcoholic fatty liver For numbered affiliations see disease (NAFLD) is modulated by genetic and end of article. the host phenotypic features—from moderate to severe obesity—may be associated with significant changes in environmental factors. ► Recent discoveries revealed the role of the Correspondence to the microbial DNA profile. Dr Silvia Sookoian, Institute Design and methods Liver tissue samples from 116 gut microbiota in human health and disease, of Medical Research A Lanari, individuals, comprising of 47 NAFLD overweight or including NAFLD. However, the impact of the University of Buenos Aires moderately obese patients, 50 NAFLD morbidly obese liver tissue microbial DNA profiling on the Faculty of Medicine, Buenos disease biology remains unknown. Aires, 10109 CABA, Argentina; patients elected for bariatric surgery and 19 controls, ssookoian@ intramed. net were analysed using high- throughput 16S rRNA gene What are the new findings? Dr Carlos Jose Pirola; sequencing. -

MICRO-ORGANISMS and RUMINANT DIGESTION: STATE of KNOWLEDGE, TRENDS and FUTURE PROSPECTS Chris Mcsweeney1 and Rod Mackie2
BACKGROUND STUDY PAPER NO. 61 September 2012 E Organización Food and Organisation des Продовольственная и cельскохозяйственная de las Agriculture Nations Unies Naciones Unidas Organization pour организация para la of the l'alimentation Объединенных Alimentación y la United Nations et l'agriculture Наций Agricultura COMMISSION ON GENETIC RESOURCES FOR FOOD AND AGRICULTURE MICRO-ORGANISMS AND RUMINANT DIGESTION: STATE OF KNOWLEDGE, TRENDS AND FUTURE PROSPECTS Chris McSweeney1 and Rod Mackie2 The content of this document is entirely the responsibility of the authors, and does not necessarily represent the views of the FAO or its Members. 1 Commonwealth Scientific and Industrial Research Organisation, Livestock Industries, 306 Carmody Road, St Lucia Qld 4067, Australia. 2 University of Illinois, Urbana, Illinois, United States of America. This document is printed in limited numbers to minimize the environmental impact of FAO's processes and contribute to climate neutrality. Delegates and observers are kindly requested to bring their copies to meetings and to avoid asking for additional copies. Most FAO meeting documents are available on the Internet at www.fao.org ME992 BACKGROUND STUDY PAPER NO.61 2 Table of Contents Pages I EXECUTIVE SUMMARY .............................................................................................. 5 II INTRODUCTION ............................................................................................................ 7 Scope of the Study ........................................................................................................... -
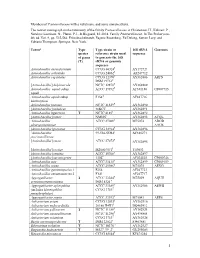
Members of Pasteurellaceae with a Valid Name and Some Unnamed Taxa
Members of Pasteurellaceae with a valid name and some unnamed taxa. The newest monograph on the taxonomy of the family Pasteurellaceae is Christensen, H., Kuhnert, P., Nørskov-Lauritsen, N., Planet, P.J., & Bisgaard, M. 2014. Family Pasteurellaceae. In The Prokaryotes 4th ed. Vol. 9, pp. 535-564. Erko Stackebrandt, Eugene Rosenberg, Ed Delong, Steven Lory, and Fabiano Thompson. Springer, New York. Taxona Type Type strain or 16S rRNA Genomes species reference strain used sequence of genus to generate the 16S (T) rRNA or genomic sequence Actinobacillus anseriformium CCUG 60324T AY172727 Actinobacillus arthritidis CCUG 24862T AF247712 Actinobacillus capsulatus CCUG 12396T AY362886 ARFN DSM 19761T [Actinobacillus] delphinicola NCTC 12870T AY362889 Actinobacillus equuli subsp. ATCC 19392T AF381186 CP007715 equuli Actinobacillus equuli subsp. F154T AF247716 haemolyticus Actinobacillus hominis NCTC 11529T AY362890 [Actinobacillus] indolicus 46KC2T AY362891 Actinobacillus lignieresii T NCTC 4189T AY362892 [Actinobacillus] minor NM305T AY362893 ACQL Actinobacillus ATCC 27088T M75074 ADOD pleuropneumoniae AACK [Actinobacillus] porcinus CCUG 38924T AY362896 ‘Actinobacillus 99-536-55H-F AF486274 porcitonsillarum’ T [Actinobacillus] rossii ATCC 27072 AY362895 [Actinobacillus] scotiae M2000/95/1T Y09653 [Actinobacillus] seminis ATCC 15768T AY362897 [Actinobacillus] succinogenes 130ZT AF024525 CP000746 Actinobacillus suis ATCC 33415T AY362899 CP009159 Actinobacillus ureae ATCC 25986T M75075 AEVG Actinobacillus genomospecies 1 F264 AF247723 Actinobacillus -

Natural Transformation As a Mechanism of Horizontal Gene Transfer in Aliarcobacter Butzleri
pathogens Article Natural Transformation as a Mechanism of Horizontal Gene Transfer in Aliarcobacter butzleri Marina Bonifácio 1, Cristiana Mateus 1, Ana R. Alves 1, Emanuel Maldonado 2 , Ana P. Duarte 1,2 , Fernanda Domingues 1,Mónica Oleastro 3 and Susana Ferreira 1,* 1 CICS-UBI-Health Sciences Research Centre, University of Beira Interior, 6201-506 Covilhã, Portugal; [email protected] (M.B.); [email protected] (C.M.); [email protected] (A.R.A.); [email protected] (A.P.D.); [email protected] (F.D.) 2 C4-UBI-Cloud Computing Competence Centre, University of Beira Interior, 6200-284 Covilhã, Portugal; [email protected] 3 National Reference Laboratory for Gastrointestinal Infections, Department of Infectious Diseases, National Institute of Health Dr. Ricardo Jorge, 1649-016 Lisbon, Portugal; [email protected] * Correspondence: [email protected] Abstract: Aliarcobacter butzleri is an emergent enteropathogen, showing high genetic diversity, which likely contributes to its adaptive capacity to different environments. Whether natural transforma- tion can be a mechanism that generates genetic diversity in A. butzleri is still unknown. In the present study, we aimed to establish if A. butzleri is naturally competent for transformation and to investigate the factors influencing this process. Two different transformation procedures were tested using exogenous and isogenic DNA containing antibiotic resistance markers, and different external conditions influencing the process were evaluated. The highest number of transformable Citation: Bonifácio, M.; Mateus, C.; A. butzleri strains were obtained with the agar transformation method when compared to the biphasic Alves, A.R.; Maldonado, E.; Duarte, system (65% versus 47%). -

Pseudomonas Aeruginosa-Derived Rhamnolipids and Other Detergents Modulate Colony Morphotype and Motility in the Burkholderia Cepacia Complex
Dartmouth College Dartmouth Digital Commons Dartmouth Scholarship Faculty Work 4-16-2017 Pseudomonas Aeruginosa-Derived Rhamnolipids and Other Detergents Modulate Colony Morphotype and Motility in the Burkholderia Cepacia Complex Steve P. Bernier McMaster University Courtney Hum McMaster University Xiang Li McMaster University George A. O'Toole Dartmouth College Nathan A. Magarvey McMaster University See next page for additional authors Follow this and additional works at: https://digitalcommons.dartmouth.edu/facoa Part of the Medicine and Health Sciences Commons Dartmouth Digital Commons Citation Bernier, Steve P.; Hum, Courtney; Li, Xiang; O'Toole, George A.; Magarvey, Nathan A.; and Surette, Michael G., "Pseudomonas Aeruginosa-Derived Rhamnolipids and Other Detergents Modulate Colony Morphotype and Motility in the Burkholderia Cepacia Complex" (2017). Dartmouth Scholarship. 3135. https://digitalcommons.dartmouth.edu/facoa/3135 This Article is brought to you for free and open access by the Faculty Work at Dartmouth Digital Commons. It has been accepted for inclusion in Dartmouth Scholarship by an authorized administrator of Dartmouth Digital Commons. For more information, please contact [email protected]. Authors Steve P. Bernier, Courtney Hum, Xiang Li, George A. O'Toole, Nathan A. Magarvey, and Michael G. Surette This article is available at Dartmouth Digital Commons: https://digitalcommons.dartmouth.edu/facoa/3135 RESEARCH ARTICLE crossm Pseudomonas aeruginosa-Derived Rhamnolipids and Other Detergents Modulate