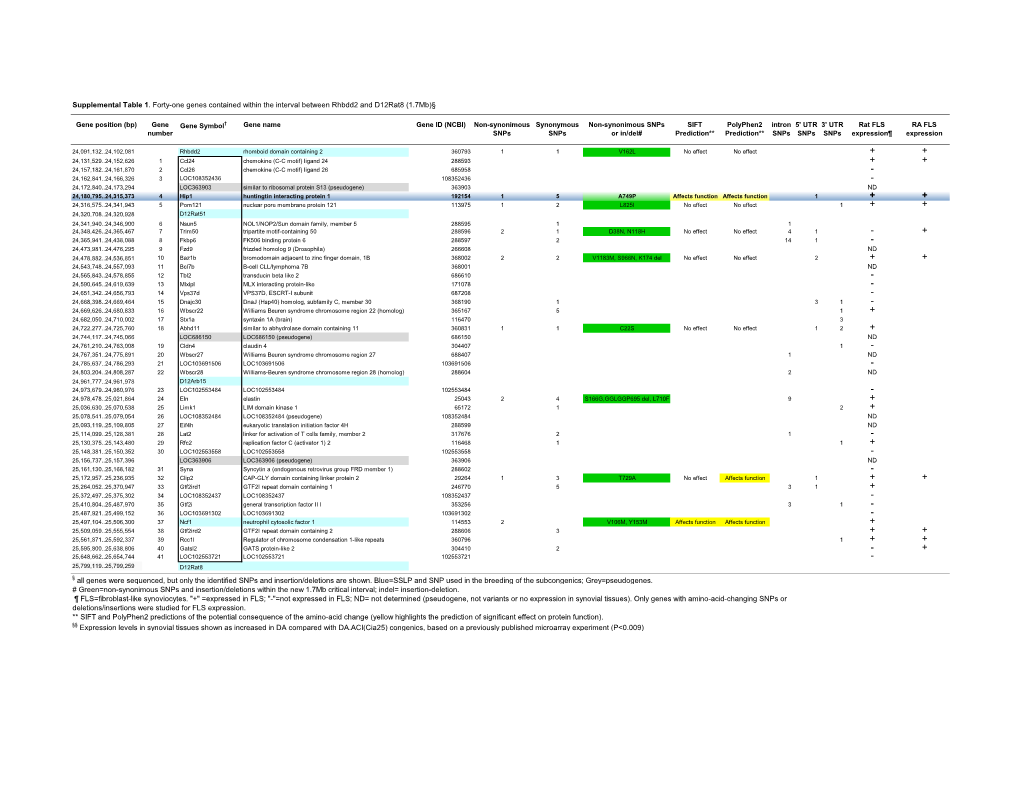
Genes Contained Within the Interval Between Rhbdd2 and D12rat8 (1.7Mb)§
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

RFC2 Antibody
Product Datasheet RFC2 Antibody Catalog No: #43122 Orders: [email protected] Description Support: [email protected] Product Name RFC2 Antibody Host Species Rabbit Clonality Polyclonal Purification Antigen affinity purification. Applications WB Species Reactivity Hu Specificity The antibody detects endogenous levels of total RFC2 protein. Immunogen Type peptide Immunogen Description Synthetic peptide of human RFC2 Target Name RFC2 Other Names RFC40 Accession No. Swiss-Prot#: P35250Gene ID: 5982 Calculated MW 39kd Concentration 3.5mg/ml Formulation Rabbit IgG in pH7.4 PBS, 0.05% NaN3, 40% Glycerol. Storage Store at -20°C Application Details Western blotting: 1:200-1:1000 Immunohistochemistry: 1:30-1:150 Images Gel: 10%SDS-PAGE Lysate: 40 µg Lane: Human liver cancer tissue Primary antibody: 1/500 dilution Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution Exposure time: 20 seconds Background This gene encodes a member of the activator 1 small subunits family. The elongation of primed DNA templates by DNA polymerase delta and epsilon requires the action of the accessory proteins, proliferating cell nuclear antigen (PCNA) and replication factor C (RFC). Replication factor C, also called activator 1, is a protein complex consisting of five distinct subunits. This gene encodes the 40 kD subunit, which has been shown to be responsible for binding ATP and may help promote cell survival. Disruption of this gene is associated with Williams syndrome. Alternatively spliced transcript variants Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 encoding distinct isoforms have been described. A pseudogene of this gene has been defined on chromosome 2. -

Program Nr: 1 from the 2004 ASHG Annual Meeting Mutations in A
Program Nr: 1 from the 2004 ASHG Annual Meeting Mutations in a novel member of the chromodomain gene family cause CHARGE syndrome. L.E.L.M. Vissers1, C.M.A. van Ravenswaaij1, R. Admiraal2, J.A. Hurst3, B.B.A. de Vries1, I.M. Janssen1, W.A. van der Vliet1, E.H.L.P.G. Huys1, P.J. de Jong4, B.C.J. Hamel1, E.F.P.M. Schoenmakers1, H.G. Brunner1, A. Geurts van Kessel1, J.A. Veltman1. 1) Dept Human Genetics, UMC Nijmegen, Nijmegen, Netherlands; 2) Dept Otorhinolaryngology, UMC Nijmegen, Nijmegen, Netherlands; 3) Dept Clinical Genetics, The Churchill Hospital, Oxford, United Kingdom; 4) Children's Hospital Oakland Research Institute, BACPAC Resources, Oakland, CA. CHARGE association denotes the non-random occurrence of ocular coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies and deafness (OMIM #214800). Almost all patients with CHARGE association are sporadic and its cause was unknown. We and others hypothesized that CHARGE association is due to a genomic microdeletion or to a mutation in a gene affecting early embryonic development. In this study array- based comparative genomic hybridization (array CGH) was used to screen patients with CHARGE association for submicroscopic DNA copy number alterations. De novo overlapping microdeletions in 8q12 were identified in two patients on a genome-wide 1 Mb resolution BAC array. A 2.3 Mb region of deletion overlap was defined using a tiling resolution chromosome 8 microarray. Sequence analysis of genes residing within this critical region revealed mutations in the CHD7 gene in 10 of the 17 CHARGE patients without microdeletions, including 7 heterozygous stop-codon mutations. -

Gene Expression Is Related to Parental Origin and Regional Coordinate Control
Journal of Human Genetics (2009) 54, 193–198 & 2009 The Japan Society of Human Genetics All rights reserved 1434-5161/09 $32.00 www.nature.com/jhg ORIGINAL ARTICLE William’s syndrome: gene expression is related to parental origin and regional coordinate control Jeremy C Collette1, Xiao-Ning Chen1, Debra L Mills2, Albert M Galaburda3, Allan L Reiss4, Ursula Bellugi5 and Julie R Korenberg1,6 William’s syndrome (WS) features a spectrum of neurocognitive and behavioral abnormalities due to a rare 1.5 MB deletion that includes about 24–28 genes on chromosome band 7q11.23. Study of the expression of these genes from the single normal copy provides an opportunity to elucidate the genetic and epigenetic controls on these genes as well as their roles in both WS and normal brain development and function. We used quantitative RT-PCR to determine the transcriptional level of 14 WS gene markers in a cohort of 77 persons with WS and 48 normal controls. Results reported here: (1) show that the expression of the genes deleted in WS is decreased in some but not all cases, (2) demonstrate that the parental origin of the deletion contributes to the level of expression of GTF2I independently of age and gender and (3) indicate that the correlation of expression between GTF2I and some other genes in the WS region differs in WS subjects and normal controls, which in turn points toward a regulatory role for this gene. Interspecies comparisons suggest GTF2I may play a key role in normal brain development. Journal of Human Genetics (2009) 54, 193–198; doi:10.1038/jhg.2009.5; published online 13 March 2009 Keywords: William’s syndrome; gene expression; RT-PCR; parental origin; GTF2I INTRODUCTION As an approach toward understanding the role of the deleted genes William’s syndrome (WS) is a neurogenetic disorder affecting human in WS, we have characterized WS subjects according to genetic, social/ development and adult cognition. -

Structure of the Human Clamp Loader Reveals an Autoinhibited Conformation of a Substrate-Bound AAA+ Switch
Structure of the human clamp loader reveals an autoinhibited conformation of a substrate-bound AAA+ switch Christl Gaubitza,1, Xingchen Liua,b,1, Joseph Magrinoa,b, Nicholas P. Stonea, Jacob Landecka,b, Mark Hedglinc, and Brian A. Kelcha,2 aDepartment of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester MA 01605; bGraduate School of Biomedical Sciences, University of Massachusetts Medical School, Worcester MA 01605; and cDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802 Edited by Michael E. O’Donnell, HHMI and Rockefeller University, New York, NY, and approved July 27, 2020 (received for review April 20, 2020) DNA replication requires the sliding clamp, a ring-shaped protein areflexia syndrome (15), Hutchinson–Gilford progeria syn- complex that encircles DNA, where it acts as an essential cofactor drome (16), and in the replication of some viruses (17–19). It for DNA polymerases and other proteins. The sliding clamp needs is unknown whether loading by RFC contributes to PARD to be opened and installed onto DNA by a clamp loader ATPase of disease. the AAA+ family. The human clamp loader replication factor C Clamp loaders are members of the AAA+ family of ATPases (RFC) and sliding clamp proliferating cell nuclear antigen (PCNA) (ATPases associated with various cellular activities), a large are both essential and play critical roles in several diseases. De- protein family that uses the chemical energy of adenosine 5′- spite decades of study, no structure of human RFC has been re- triphosphate (ATP) to generate mechanical force (20). Most solved. Here, we report the structure of human RFC bound to AAA+ proteins form hexameric motors that use an undulating PCNA by cryogenic electron microscopy to an overall resolution ∼ spiral staircase mechanism to processively translocate a substrate of 3.4 Å. -

Identification and Characterization of a Novel Non-Homologous End Joining Factor MRI
Washington University in St. Louis Washington University Open Scholarship Arts & Sciences Electronic Theses and Dissertations Arts & Sciences Spring 5-15-2020 Identification and Characterization of a Novel Non-homologous End Joining Factor MRI Putzer Joseph Hung Washington University in St. Louis Follow this and additional works at: https://openscholarship.wustl.edu/art_sci_etds Part of the Allergy and Immunology Commons, Immunology and Infectious Disease Commons, and the Medical Immunology Commons Recommended Citation Hung, Putzer Joseph, "Identification and Characterization of a Novel Non-homologous End Joining Factor MRI" (2020). Arts & Sciences Electronic Theses and Dissertations. 2201. https://openscholarship.wustl.edu/art_sci_etds/2201 This Dissertation is brought to you for free and open access by the Arts & Sciences at Washington University Open Scholarship. It has been accepted for inclusion in Arts & Sciences Electronic Theses and Dissertations by an authorized administrator of Washington University Open Scholarship. For more information, please contact [email protected]. WASHINGTON UNIVERSITY IN ST. LOUIS Division of Biology and Biomedical Sciences Immunology Dissertation Examination Committee: Barry Sleckman, Chair Gaya Amarasinghe Brian Edelson Takeshi Egawa Nima Mosammaparast Kenneth Murphy Sheila Stewart Identification and Characterization of a Novel Non-homologous End Joining Factor MRI by Putzer Joseph Hung A dissertation presented to The Graduate School of Washington University in partial fulfillment of the requirements -

The Nuclear Localization Pattern and Interaction Partners of GTF2IRD1 Demonstrate a Role in Chromatin Regulation
Hum Genet DOI 10.1007/s00439-015-1591-0 ORIGINAL INVESTIGATION The nuclear localization pattern and interaction partners of GTF2IRD1 demonstrate a role in chromatin regulation Paulina Carmona‑Mora1 · Jocelyn Widagdo2 · Florence Tomasetig1 · Cesar P. Canales1 · Yeojoon Cha1 · Wei Lee1 · Abdullah Alshawaf3 · Mirella Dottori3 · Renee M. Whan4 · Edna C. Hardeman1 · Stephen J. Palmer1 Received: 11 February 2015 / Accepted: 4 August 2015 © Springer-Verlag Berlin Heidelberg 2015 Abstract GTF2IRD1 is one of the three members of the mostly involved in chromatin modification and transcrip- GTF2I gene family, clustered on chromosome 7 within a tional regulation, whilst others indicate an unexpected role 1.8 Mb region that is prone to duplications and deletions in connection with the primary cilium. Mapping of the sites in humans. Hemizygous deletions cause Williams–Beuren of protein interaction also indicates key features regarding syndrome (WBS) and duplications cause WBS duplica- the evolution of the GTF2IRD1 protein. These data provide tion syndrome. These copy number variations disturb a a visual and molecular basis for GTF2IRD1 nuclear func- variety of developmental systems and neurological func- tion that will lead to an understanding of its role in brain, tions. Human mapping data and analyses of knockout mice behaviour and human disease. show that GTF2IRD1 and GTF2I underpin the craniofacial abnormalities, mental retardation, visuospatial deficits and Abbreviations hypersociability of WBS. However, the cellular role of the hESC Human embryonic stem cells GTF2IRD1 protein is poorly understood due to its very PLA Proximity ligation assay low abundance and a paucity of reagents. Here, for the first STED Stimulated emission depletion time, we show that endogenous GTF2IRD1 has a punctate WBS Williams–Beuren syndrome pattern in the nuclei of cultured human cell lines and neu- Y2H Yeast two-hybrid rons. -

The Contribution of CLIP2 Haploinsufficiency to the Clinical
REPORT The Contribution of CLIP2 Haploinsufficiency to the Clinical Manifestations of the Williams-Beuren Syndrome Geert Vandeweyer,1 Nathalie Van der Aa,1 Edwin Reyniers,1 and R. Frank Kooy1,* Williams-Beuren syndrome is a rare contiguous gene syndrome, characterized by intellectual disability, facial dysmorphisms, connec- tive-tissue abnormalities, cardiac defects, structural brain abnormalities, and transient infantile hypercalcemia. Genes lying telomeric to RFC2, including CLIP2, GTF2I and GTF2IRD1, are currently thought to be the most likely major contributors to the typical Williams syndrome cognitive profile, characterized by a better-than-expected auditory rote-memory ability, a relative sparing of language capa- bilities, and a severe visual-spatial constructive impairment. Atypical deletions in the region have helped to establish genotype-pheno- type correlations. So far, however, hardly any deletions affecting only a single gene in the disease region have been described. We present here two healthy siblings with a pure, hemizygous deletion of CLIP2. A putative role in the cognitive and behavioral abnormalities seen in Williams-Beuren patients has been suggested for this gene on the basis of observations in a knock-out mouse model. The presented siblings did not show any of the clinical features associated with the syndrome. Cognitive testing showed an average IQ for both and no indication of the Williams syndrome cognitive profile. This shows that CLIP2 haploinsufficiency by itself does not lead to the physical or cognitive characteristics of the Williams-Beuren syndrome, nor does it lead to the Williams syndrome cognitive profile. Although contri- bution of CLIP2 to the phenotype cannot be excluded when it is deleted in combination with other genes, our results support the hypothesis that GTF2IRD1 and GTF2I are the main genes causing the cognitive defects associated with Williams-Beuren syndrome. -

GTF2IRD1 Rabbit Polyclonal Antibody – AP06760PU-N | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AP06760PU-N GTF2IRD1 Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: Western blot: 1/500-1/1000. Reactivity: Human, Mouse, Rat Host: Rabbit Clonality: Polyclonal Immunogen: Synthetic peptide, corresponding to amino acids 63-112 of Human WBSCR11. Specificity: This antibody detects endogenous levels of WBSCR11 protein. (region surrounding Lys94) Formulation: Phosphate buffered saline (PBS), pH~7.2 State: Aff - Purified State: Liquid purified Ig fraction (> 95% pure by SDS-PAGE) Preservative: 0.05% Sodium Azide Concentration: 1.0 mg/ml Purification: Affinity Chromatography using epitope-specific immunogen Conjugation: Unconjugated Storage: Store undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Predicted Protein Size: ~106 kDa Database Link: Entrez Gene 9569 Human Q9UHL9 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 GTF2IRD1 Rabbit Polyclonal Antibody – AP06760PU-N Background: Williams-Beuren syndrome (WBS) is a developmental disorder caused by the hemizygous microdeletion on chromosome 7q11.23. WBS is an autosomal dominant genetic condition that is characterized by physical, cognitive and behavioral traits. The physical traits associated with WBS include facial dysmorphology, vascular stenoses, growth deficiencies, dental anomalies and neurologic and musculoskeletal abnormalities. -

Supplemental Figures
Supplemental Figures Supplemental figure legends Figure S1 | Testing the pre-clustering heuristic. (A) (Left) Default, unsupervised heuristic sets a cut of 7% of the total dendrogram depth, which results in 52 pre-clusters. (Right) The numerical model calculated using the 52 pre-clusters. Xc1 and Xc2 represent the expression (in a binned UMIs grid) of a given gene X in two cells c1 and c2 belonging to the same pre-cluster. The cumulative distribution plot estimates the frequency, hence likelihood, of an expression change. (B) (Left) Forcing a cut of only 4% creates 1152 pre-clusters, more than 20-fold increase compared to the default 7% depth. Also, given the reduction of the average cluster size and the consequent reduction of possible intra-cluster pair-wise comparison, the number of data points used to fit the model decreases of more than 5-fold compared to default 7% cut (from 3.79E+9 to 6.56E+8). (Right) Despite this, the difference between the numerical model of 4% cut and 7% cut is marginal. (C) (Left) Forcing a cut of 20% creates only 9 pre-clusters, which is less than the number of final clusters (in this case, 11) and therefore represents a miscalculated configuration. Still the difference between the numerical model of 20% cut and 7% cut is marginal (right). (D) Also switching from Pearson to Spearman correlation is associated with neglectable differences in the numerical model. (E) (Top) Number of pre-clusters associated with the different cutting depths, correlations metrics (Pearson, Spearman) or linkage metrics (complete or Weighted average distance, WPGMA, instead of default Ward’s). -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2-