Biological Therapy in Systemic Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Vivo Imaging of Tgfβ Signalling Components Using Positron
REVIEWS Drug Discovery Today Volume 24, Number 12 December 2019 Reviews KEYNOTE REVIEW In vivo imaging of TGFb signalling components using positron emission tomography 1 1 2 Lonneke Rotteveel Lonneke Rotteveel , Alex J. Poot , Harm Jan Bogaard , received her MSc in drug 3 1 discovery and safety at the Peter ten Dijke , Adriaan A. Lammertsma and VU University in 1 Amsterdam. She is Albert D. Windhorst currently finishing her PhD at the VU University 1 Department of Radiology and Nuclear Medicine, Amsterdam UMC, location VUmc, Amsterdam, The Netherlands Medical Center (VUmc) 2 under the supervision of A. Pulmonary Medicine, Institute for Cardiovascular Research, Amsterdam UMC, location VUmc, Amsterdam, The Netherlands D. Windhorst and Adriaan A. Lammertsma. Her 3 research interest is on the development of positron Department of Cell and Chemical Biology, Oncode Institute, Leiden University Medical Center, Leiden, The emission tomography (PET) tracers that target Netherlands selectively the activin receptor-like kinase 5 in vitro and in vivo. Alex J. Poot obtained his The transforming growth factor b (TGFb) family of cytokines achieves PhD in medicinal chemistry homeostasis through a careful balance and crosstalk with complex from Utrecht University. As postdoctoral researcher at signalling pathways. Inappropriate activation or inhibition of this pathway the VUmc, Amsterdam, he and mutations in its components are related to diseases such as cancer, developed radiolabelled anticancer drugs for PET vascular diseases, and developmental disorders. Quantitative imaging of imaging. In 2014, he accepted a research expression levels of key regulators within this pathway using positron fellowship from Memorial Sloan Kettering Cancer 13 emission tomography (PET) can provide insights into the role of this Center, New York to develop C-labelled probes for tumour metabolism imaging with magnetic resonance in vivo pathway , providing information on underlying pathophysiological imaging (MRI). -

Treatment of Diffuse Cutaneous Systemic Sclerosis with Hyperimmune Caprine Serum
University College London (UCL) MD (Res) Degree Treatment of Diffuse Cutaneous Systemic Sclerosis with Hyperimmune Caprine Serum DR. NIAMH PATRICIA QUILLINAN 1 Declaration I, Niamh Patricia Quillinan, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed: ___________________________________ Date: 17/03/2016 2 Abstract Systemic sclerosis (SSc) is a multisystem autoimmune rheumatic disorder with high morbidity and the highest case specific mortality of the rheumatic diseases. There is no currently approved unequivocally effective treatment for SSc and therefore there is a huge unmet medical need for novel and effective therapies. Hyperimmune caprine serum (HCS) is a goat serum extract derivative produced from goats vaccinated with a detergent-inactivated HIV viral lysate. It contains caprine immunoglobulins and small molecular weight proteins as well as a CRH, α-2 macroglobulin (α-2M) and lipoprotein-related peptide-1 complex. In this thesis we explore the hypothesis that hyperimmune caprine serum improves skin and other measures of disease severity in established dcSSc by modulating immunological function that determines persistence of clinical disease. This hypothesis is explored through 1) a prospective clinical trial, 2) long-term clinical use and 3) detailed assessment of serum growth factors and cytokines, as well as established and exploratory markers of disease. The primary objective of the clinical trial was to explore safety and tolerability of HCS in established diffuse cutaneous systemic sclerosis (dcSSc). Secondary objectives included assessment of potential efficacy and biological activity and exploration of candidate biomarkers. There were no safety concerns and frequency of adverse events was not different between HCS and placebo group. -

Clinical Research Newsletter for Colleagues in the Community
Clinical Research Newsletter for Colleagues in the Community Welcome to the Winter 2017 issue of the Stanford Cancer surgery, robotic surgery, stereotactic radiosurgery such as Institute Clinical Research Newsletter. This quarterly CyberKnife®, microvascular reconstruction, intraoperative publication is designed to inform our colleagues in the radiation therapy (IORT), along with new chemotherapy trials. medical community, and especially physicians who are The Stanford Cancer Institute Neuro-Oncology Program considering treatment options for their patients with offers Phase I through III trials as well as multidisciplinary, cancer, about current clinical trials available at the Stanford collaborative evaluation and treatment of patients with tumors Cancer Institute, a National Cancer Institute designated of the nervous system. This includes, but is not restricted Comprehensive Cancer Center. Many of these trials provide to; brain metastases, leptomeningeal cancer, glioblastomas access to novel therapies including new “targeted” agents, and less aggressive gliomas, benign brain and spinal tumors, often not available in the community. and base of brain neoplasms including pituitary disorders. As leaders of the Stanford Head and Neck Cancer Care Clinical trials have focused on vaccine therapy, antibody Program, we are delighted to introduce this edition of the therapy, novel chemotherapy agents, radiation sensitizers, newsletter, as it focuses on our Neuro-Oncology, Thoracic novel radiation therapy, and radiosurgery techniques. Oncology, and Developmental Therapeutics programs. The Developmental Therapeutics Program conducts Each of these programs offers cutting-edge clinical trials pharmacokinetic and pharmacodynamic driven first-in- for patients with tumors that can be challenging to treat human trials tailored to make early, informed decisions with current routine care. Weekly multidisciplinary tumor regarding the suitability of novel molecular agents for boards are available for each program. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub
US 20170172932A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub. Date: Jun. 22, 2017 (54) EARLY CANCER DETECTION AND A 6LX 39/395 (2006.01) ENHANCED IMMUNOTHERAPY A61R 4I/00 (2006.01) (52) U.S. Cl. (71) Applicant: Gholam A. Peyman, Sun City, AZ CPC .......... A61K 9/50 (2013.01); A61K 39/39558 (US) (2013.01); A61K 4I/0052 (2013.01); A61 K 48/00 (2013.01); A61K 35/17 (2013.01); A61 K (72) Inventor: sham A. Peyman, Sun City, AZ 35/15 (2013.01); A61K 2035/124 (2013.01) (21) Appl. No.: 15/143,981 (57) ABSTRACT (22) Filed: May 2, 2016 A method of therapy for a tumor or other pathology by administering a combination of thermotherapy and immu Related U.S. Application Data notherapy optionally combined with gene delivery. The combination therapy beneficially treats the tumor and pre (63) Continuation-in-part of application No. 14/976,321, vents tumor recurrence, either locally or at a different site, by filed on Dec. 21, 2015. boosting the patient’s immune response both at the time or original therapy and/or for later therapy. With respect to Publication Classification gene delivery, the inventive method may be used in cancer (51) Int. Cl. therapy, but is not limited to such use; it will be appreciated A 6LX 9/50 (2006.01) that the inventive method may be used for gene delivery in A6 IK 35/5 (2006.01) general. The controlled and precise application of thermal A6 IK 4.8/00 (2006.01) energy enhances gene transfer to any cell, whether the cell A 6LX 35/7 (2006.01) is a neoplastic cell, a pre-neoplastic cell, or a normal cell. -

Recommended INN: List 63
WHO Drug Information, Vol. 24, No. 1, 2010 Recommended INN: List 63 International Nonproprietary Names for Pharmaceutical Substances (INN) RECOMMENDED International Nonproprietary Names: List 63 Notice is hereby given that, in accordance with paragraph 7 of the Procedure for the Selection of Recommended International Nonproprietary Names for Pharmaceutical Substances [Off. Rec. Wld Health Org., 1955, 60, 3 (Resolution EB15.R7); 1969, 173, 10 (Resolution EB43.R9); Resolution EB115.R4 (EB115/2005/ERC/1)], the following names are selected as Recommended International Nonproprietary Names. The inclusion of a name in the lists of Recommended International Nonproprietary Names does not imply any recommendation of the use of the substance in medicine or pharmacy. Lists of Proposed (1–101) and Recommended (1–62) International Nonproprietary Names can be found in Cumulative List No. 13, 2009 (available in CD-ROM only). Dénominations communes internationales des Substances pharmaceutiques (DCI) Dénominations communes internationales RECOMMANDÉES: Liste 63 Il est notifié que, conformément aux dispositions du paragraphe 7 de la Procédure à suivre en vue du choix de Dénominations communes internationales recommandées pour les Substances pharmaceutiques [Actes off. Org. mond. Santé, 1955, 60, 3 (résolution EB15.R7); 1969, 173, 10 (résolution EB43.R9); résolution EB115.R4 (EB115/2005/ERC/1)] les dénominations ci-dessous sont choisies par l’Organisation mondiale de la Santé en tant que dénominations communes internationales recommandées. L’inclusion d’une dénomination dans les listes de DCI recommandées n’implique aucune recommandation en vue de l’utilisation de la substance correspondante en médecine ou en pharmacie. On trouvera d’autres listes de Dénominations communes internationales proposées (1–101) et recommandées (1–62) dans la Liste récapitulative No. -
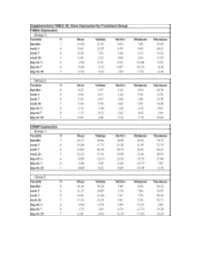
Fresolimumab Suppl Methods
Supplementary Figure 1. Fresolimumab treated patients show decreased biomarkers and skin scores. Graphs show thrombopondin-1 (THBS1, panels a and b) and cartilage oligomeric protein (COMP, panels c and d) gene expression, and MRSS (panels e and f) from study patients in group 1, receiving two doses of 1 mg/kg fresolimumab, (a, c, and e) and group 2, receiving one dose of 5 mg/kg fresolimumab (b, d and f). Line graphs show changes in individual patients over time. A B C Supplementary Figure 2: Biomarker gene expression correlates with the local skin score. The local forearm skin score correlates highly with the MRSS (panel a), as well as THBS1 (panel b) and COMP (panel c) gene expression. Supplementary Figure 3. Autoantibody levels measured at baseline and 11 weeks after fresolimumab treatment. Anti-RNA polymerase III levels for anti-RNA polymerase III- positive patients are shown in blue. Anti-Scl70 levels for anti-Scl70-positive patients are shown in red. Autoantibody levels in two of the four anti-Scl70-positive patients lie on top of the other two. Supplementary Figure 4. Disease duration compared to the change in MRSS 7 weeks after fresolimumab treatment. The change in MRSS after fresolimumab treatment showed no correlation with the disease duration (r2 = 0.00063) Supplementary Figure 5A. Immunohistochemical Supplementary Figure 5B. Skin collagen staining of plasminogen activator protein-1 (PAI- thickness. Based on trichrome staining, dermal 1/serpine1). Each patient is represented by a thickness was measured from the dermal-epidermal different color/style of marker. The median of the junction to subcutaneous fat. -

Epithelial–Mesenchymal Transition in Crohn's Disease
REVIEW Epithelial–mesenchymal transition in Crohn’s disease H Jiang1, J Shen1 and Z Ran1 Crohn’s disease (CD) is often accompanied by the complications of intestinal strictures and fistulas. These complications remain obstacles in CD treatment. In recent years, the importance of epithelial–mesenchymal transition in the pathogenesis of CD-associated fistulas and intestinal fibrosis has become apparent. Epithelial–mesenchymal transition refers to a dynamic change, wherein epithelial cells lose their polarity and adherence and acquire migratory function and fibroblast features. During formation of CD-associated fistulas, intestinal epithelial cells dislocate from the basement membrane and migrate to the lining of the fistula tracts, where they convert into transitional cells as a compensatory response under the insufficient wound healing condition. In CD-associated intestinal fibrosis, epithelial–mesenchymal transition may serve as a source of new fibroblasts and consequently lead to overproduction of extracellular matrix. In this review, we present current knowledge of epithelial–mesenchymal transition and its role in the pathogenesis of CD in order to highlight new therapy targets for the associated complications. INTRODUCTION and fistula formation in CD.6,7 EMT was first identified in the Therapeutic strategies for Crohn’s disease (CD) have progressed 1960s from the pioneering work of Elizabeth Hay, who remarkably in the past decade.1 From 1991 to 2014, the rate of described transformation of epithelial cells to mesenchymal hospitalization and surgical intervention for CD decreased; cells in the chick primitive streak during embryogenesis.8 however, despite the use of advanced drugs such as immuno- Generally, in EMT, epithelial cells demonstrate plasticity and modulators and biologics, the progression to complicated disease lose epithelial markers like E-cadherin and catenins. -

Targeting TGF-Β Signaling for Therapeutic Gain
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Targeting TGF-b Signaling for Therapeutic Gain Rosemary J. Akhurst Department of Anatomy and Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California 94158-9001 Correspondence: [email protected] Transforming growth factor bs (TGF-bs) are closely related ligands that have pleiotropic activity on most cell types of the body. They act through common heterotetrameric TGF-b type II and type I transmembrane dual specificity kinase receptor complexes, and the outcome of signaling is context-dependent. In normal tissue, they serve a role in maintaining homeostasis. In many diseased states, particularly fibrosis and cancer, TGF-b ligands are overexpressed and the outcome of signaling is diverted toward disease progression. There has therefore been a concerted effort to develop drugs that block TGF-b signaling for therapeutic benefit. This review will cover the basics of TGF-b signaling and its biological activities relevant to oncology, present a summary of pharmacological TGF-b blockade strategies, and give an update on preclinical and clinical trials for TGF-b blockade in a variety of solid tumor types. he three transforming growth factor bs titude of TGF-b-induced tumor promoting ef- T(TGF-bs), TGF-b1, -b2, and -b3, are closely fects modulate the tumor cells directly through related ligands that act on most cells types of the enhancement of tumor cell invasion and metas- body and have pleiotropic activities. Expression tasis, and induction and maintenance of cells of TGF-b1 is activated by tissue perturbations with tumor initiating properties, sometimes that induce cellular stress, such as cell prolifera- termed cancer stem-like cells (CSCs). -

Is There a Role for Radiation Therapy and Immunotherapy?
IS THERE A ROLE FOR RADIATION THERAPY AND IMMUNOTHERAPY? S. Lewis Cooper, M.D. Assistant Professor, Radiation Oncology Hollings Cancer Center Medical University of South Carolina (MUSC) DISCLOSURE Abney Clinical Scholar OBJECTIVES • Review mechanisms of immune escape by cancer cells • Review radiation effects on the immune system • Review current strategies to combine RT and immunotherapy • IL-2 • Dendritic cell production • Tumor antigen vaccine • CTLA-4 antibody/PD-1antibody • TLR agonist • TGF-β antibody CANCER IMMUNOEDITING CD8+ NKT CD8+ T cell cell T cell • Three phases: NK CD4+ T cell • Elimination – Innate and adaptive immune systems detect INF-γ IL-12 and destroy developing tumor before it is clinically TNF TRAIL apparent. Mφ Perforin γδ T DC CD8+ IL-12 cell T cell CD4+ INF-γ T cell • Equilibrium – The immune system maintains residual tumor cells in a functional state of dormancy. Protection Mφ CD8+ NK T cell TGF-β PD-L1 IL-6, IL-10 • Escape – Tumor cells that aquire the ability to escape Galactin-1 IDO Antigen Loss immune recognition and destruction emerge as growing MHC Loss tumors. CD8+ T cell CTLA-4 CTLA-4 PD-1 PD-1 CD8+ MDSC Treg Science . 2011;331:1565-70 T cell MECHANISMS OF ESCAPE • Loss of tumor antigen expression 1. Tumor cells that do not express strong rejection antigens. 2. Loss of MHC class 1 proteins that present these antigens 3. Loss of antigen processing function • Immunosuppresive state in the tumor microenvironment 1. Production of immunosuppresive cytokines (VGEF, TGF-β, galectin, IDO) 2. Recruitment of immunosuppressive cells (Treg, MDSCs, TAMs) Science . -

NCT# NCT02538471 WCM IRB # 1505016222 Title: LY2157299
NCT# NCT02538471 WCM IRB # 1505016222 Title: LY2157299 Monohydrate (LY2157299) and Radiotherapy in Metastatic Breast Cancer Final Approved version date: 04.21.2017 IRB approval date: May 19th, 2017. Document History Amendments Version Version Date Protocol Amendment 4 4.0 04.21.2017 Protocol Amendment 3 3.0 11.22.2016 Protocol Amendment 2 2.1 07.18.2016 Protocol Amendment 1 2.0 09.29.2015 Initial Protocol 1.0 07.10.2015 Protocol # 1505016222 IND # 126952 Version Date 04/21/2017 Co-Investigators UCLA: 2 Protocol # 1505016222 IND # 126952 Version Date 04/21/2017 Study Contact at WCM: Sharanya Chandrasekhar 3 Protocol # 1505016222 IND # 126952 Version Date 04/21/2017 Participating Sites: 1. New York-Presbyterian Weill Cornell Medicine Stich Radiation N-046 525 East 68th street, New York, NY – 10065 2. David Geffen School of Medicine at UCLA 10833 Le Conte Avenue Los Angeles, CA 90095-1714 4 Protocol # 1505016222 IND # 126952 Version Date 04/21/2017 Confidentiality Statement This document is confidential and is to be distributed for review only to investigators, potential investigators, consultants, study staff, and applicable independent ethics committees or institutional review boards. The contents of this document shall not be disclosed to others without written authorization from WCM. 5 Protocol # 1505016222 IND # 126952 Version Date 04/21/2017 List of Abbreviations All abbreviations used throughout the protocol must be defined. 1D11 Murine monoclonal antibody against TGFΒ 4T1 murine murine breast tumor model model ADL Activities of daily -

13-400 Phrma Stateclinicaltrials MN 120913.Indd
Research in Your Backyard Developing Cures, Creating Jobs PHARMACEUTICAL CLINICAL TRIALS IN Dots show locations of clinical trials in the state. MINNESOTA Executive Summary Clinical Trials in Minnesota “ Advancements in medicine would not be • Biopharmaceutical research companies have possible without individuals willing to volunteer conducted more than 2,900 clinical trials of new their time to participate in clinical studies. medicines since 1999 in collaboration with the The Minnesota Clinical Research Association state’s local research institutions, including the Clinical Research Institute in Minneapolis and (MCRA) is a collaboration of Minnesota based Plymouth, Twin Cities Clinical Research/ research institutions and professionals who are Prism Clinical Research in Brooklyn Center, committed to objectively informing the general Prism Clinical Research in St. Paul, MAPS public to the benefits and risks involved in Applied Research Center in Edina, Minnesota clinical research far beyond international and Diet Research Center/Frestedt Inc. in St. Louis domestic regulations. MCRA members believe Park, Mercy Hospital in Coon Rapids, St. Mary’s that a fully informed participant is critical to Duluth Clinic in Duluth, St. Cloud Hospital in successful clinical research outcomes.” St. Cloud, St. Paul’s Heart Clinic in St. Paul, the — Jeff Cosgrove, President University of Minnesota School of Medicine in Prism Clinical Research Minneapolis and the Mayo Clinic in Rochester. • The trials have been aimed at a wide array of diseases and medical conditions, ranging from • MCRA provides important clinical research the most debilitating chronic diseases, including education at a time when it is a challenge for cancer, pain, heart disease, obesity and diabetes, to biopharmaceutical and device companies and bothersome allergies, sinusitis, chronic obstructive local research collaborators to recruit volunteers, pulmonary disease (COPD), migraine headaches, and patients often don’t know about trials of pneumonia and rare diseases affecting small new drugs. -

Transforming Growth Factor-β in Stem Cells and Tissue Homeostasis
Bone Research www.nature.com/boneres REVIEW ARTICLE OPEN Transforming growth factor-β in stem cells and tissue homeostasis Xin Xu1, Liwei Zheng2, Quan Yuan3, Gehua Zhen4, Janet L. Crane4,5, Xuedong Zhou1 and Xu Cao4 TGF-β 1–3 are unique multi-functional growth factors that are only expressed in mammals, and mainly secreted and stored as a latent complex in the extracellular matrix (ECM). The biological functions of TGF-β in adults can only be delivered after ligand activation, mostly in response to environmental perturbations. Although involved in multiple biological and pathological processes of the human body, the exact roles of TGF-β in maintaining stem cells and tissue homeostasis have not been well-documented until recent advances, which delineate their functions in a given context. Our recent findings, along with data reported by others, have clearly shown that temporal and spatial activation of TGF-β is involved in the recruitment of stem/progenitor cell participation in tissue regeneration/remodeling process, whereas sustained abnormalities in TGF-β ligand activation, regardless of genetic or environmental origin, will inevitably disrupt the normal physiology and lead to pathobiology of major diseases. Modulation of TGF- β signaling with different approaches has proven effective pre-clinically in the treatment of multiple pathologies such as sclerosis/ fibrosis, tumor metastasis, osteoarthritis, and immune disorders. Thus, further elucidation of the mechanisms by which TGF-β is activated in different tissues/organs and how targeted cells respond in a context-dependent way can likely be translated with clinical benefits in the management of a broad range of diseases with the involvement of TGF-β.