Oxidative Folding of a Cystine Knot Protein: the Amaranthus -Amylase Inhibitor
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Thioredoxin Trx-1 Regulates the Major Oxidative Stress Response Transcription Factor, Skn-1, in Caenorhabditis Elegans
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2016 THE THIOREDOXIN TRX-1 REGULATES THE MAJOR OXIDATIVE STRESS RESPONSE TRANSCRIPTION FACTOR, SKN-1, IN CAENORHABDITIS ELEGANS Katie C. McCallum Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cellular and Molecular Physiology Commons, Medicine and Health Sciences Commons, Molecular Genetics Commons, and the Organismal Biological Physiology Commons Recommended Citation McCallum, Katie C., "THE THIOREDOXIN TRX-1 REGULATES THE MAJOR OXIDATIVE STRESS RESPONSE TRANSCRIPTION FACTOR, SKN-1, IN CAENORHABDITIS ELEGANS" (2016). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 655. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/655 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. THE THIOREDOXIN TRX-1 REGULATES THE MAJOR OXIDATIVE STRESS RESPONSE TRANSCRIPTION FACTOR, SKN-1, IN CAENORHABDITIS ELEGANS A DISSERTATION Presented to the Faculty of The University of Texas Health Science Center at Houston and The University of Texas MD Anderson Cancer Center Graduate School of Biomedical Sciences in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY by Katie Carol McCallum, B.S. -

Structure of the Thioredoxin-Fold Domain of Human Phosducin-Like
protein structure communications Acta Crystallographica Section F Structural Biology Structure of the thioredoxin-fold domain of human and Crystallization phosducin-like protein 2 Communications ISSN 1744-3091 Xiaochu Lou,a Rui Bao,a Human phosducin-like protein 2 (hPDCL2) has been identified as belonging to Cong-Zhao Zhoub and subgroup II of the phosducin (Pdc) family. The members of this family share an Yuxing Chena,b* N-terminal helix domain and a C-terminal thioredoxin-fold (Trx-fold) domain. The X-ray crystal structure of the Trx-fold domain of hPDCL2 was solved at ˚ aInstitute of Protein Research, Tongji University, 2.70 A resolution and resembled the Trx-fold domain of rat phosducin. Shanghai 200092, People’s Republic of China, Comparative structural analysis revealed the structural basis of their putative and bHefei National Laboratory for Physical functional divergence. Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei, Anhui 230027, People’s Republic of China Correspondence e-mail: [email protected] 1. Introduction The thioredoxin-fold (Trx-fold) protein-structure classification (SCOP; Received 21 October 2008 http://scop.mrc-lmb.cam.ac.uk; Murzin et al., 1995) was characterized Accepted 11 November 2008 based on the structure of Escherichia coli Trx1 (Holmgren et al., 1975). The classic Trx fold consists of a single domain with a central PDB Reference: thioredoxin-fold domain of five-stranded mixed -sheet flanked by two -helices on each side. human phosducin-like protein 2, 3evi, r3evisf. The secondary-structure elements are arranged in the order - , with 4 antiparallel to the other strands. -

SMALL-MOLECULE CATALYSIS of NATIVE DISULFIDE BOND FORMATION in PROTEINS by Kenneth J. Woycechowsky a Dissenation Submitted in Pa
SMALL-MOLECULE CATALYSIS OF NATIVE DISULFIDE BOND FORMATION IN PROTEINS by Kenneth J. Woycechowsky A dissenation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biochemistry) at the UNIVERSITY OF WISCONSIN-MADISON 2002 A dissertation entitled Small-Molecule Ca~alysis of Na~ive Disulfide Bond Formation in Proteins submitted to the Graduate School of the University of Wisconsin-Madison in partial fulfillment of the requirements for the degree of Doctor of Philosophy by Kenneth J. Woycechowsky Date of Final Oral Examination: 19 September 2002 Month & Year Degree to be awarded: December 2002 May August **** .... **************** .... ** ...... ************** ........... "1"II_Ir'IIII;;IISIJii:iAh;;;;;?f~D~i:s:sertation Readers: Signature, Dean of Graduate School , 1 Abstract Native disulfide bond formation is essential for the folding of many proteins. The enzyme protein disulfide isomerase (POI) catalyzes native disulfide bond formation using a Cys-Gly-His Cys active site. The active-site properties of POI (thiol pK. =6.7 and disulfide E;O' =-180 mY) are critical for efficient catalysis. This Dissertation describes the design, synthesis, and characterization of small-molecule catalysts that mimic the active-site properties of POI. Chapter Two describes a small-molecule dithiol that has disulfide bond isomerization activity both in vitro and in vivo. The dithiol trans-I,2-bis(mercaptoacetamido)cyclohexane (BMC) has a thiol pKa value of 8.3 and a disulfide E;O' value of -240 mV.ln vitro, BMC increases the folding efficiency of disulfide--scrambled ribonuclease A (sRNase A). Addition of BMC to the growth medium of yeast cells causes an increase in the heterologous secretion of Schizosaccharomyces pombe acid phosphatase. -

A Shape-Shifting Redox Foldase Contributes to Proteus Mirabilis Copper Resistance
ARTICLE Received 7 Oct 2016 | Accepted 24 May 2017 | Published 19 Jul 2017 DOI: 10.1038/ncomms16065 OPEN A shape-shifting redox foldase contributes to Proteus mirabilis copper resistance Emily J. Furlong1, Alvin W. Lo2,3, Fabian Kurth1,w, Lakshmanane Premkumar1,2,w, Makrina Totsika2,3,w, Maud E.S. Achard2,3,w, Maria A. Halili1, Begon˜a Heras4, Andrew E. Whitten1,w, Hassanul G. Choudhury1,w, Mark A. Schembri2,3 & Jennifer L. Martin1,5 Copper resistance is a key virulence trait of the uropathogen Proteus mirabilis. Here we show that P. mirabilis ScsC (PmScsC) contributes to this defence mechanism by enabling swarming in the presence of copper. We also demonstrate that PmScsC is a thioredoxin-like disulfide isomerase but, unlike other characterized proteins in this family, it is trimeric. PmScsC trimerization and its active site cysteine are required for wild-type swarming activity in the presence of copper. Moreover, PmScsC exhibits unprecedented motion as a consequence of a shape-shifting motif linking the catalytic and trimerization domains. The linker accesses strand, loop and helical conformations enabling the sampling of an enormous folding land- scape by the catalytic domains. Mutation of the shape-shifting motif abolishes disulfide isomerase activity, as does removal of the trimerization domain, showing that both features are essential to foldase function. More broadly, the shape-shifter peptide has the potential for ‘plug and play’ application in protein engineering. 1 Institute for Molecular Bioscience, University of Queensland, St Lucia, Queensland 4072, Australia. 2 School of Chemistry and Molecular Biosciences, University of Queensland, St Lucia, Queensland 4072, Australia. -

A Tri-Gram Based Feature Extraction Technique Using Linear Probabilities of Position Specific Scoring Matrix for Protein Fold Recognition
A Tri-Gram Based Feature Extraction Technique Using Linear Probabilities of Position Specific Scoring Matrix for Protein Fold Recognition Author Paliwal, Kuldip K, Sharma, Alok, Lyons, James, Dehzangi, Abdollah Published 2014 Journal Title IEEE Transactions on NanoBioscience DOI https://doi.org/10.1109/TNB.2013.2296050 Copyright Statement © 2014 IEEE. Personal use of this material is permitted. Permission from IEEE must be obtained for all other uses, in any current or future media, including reprinting/republishing this material for advertising or promotional purposes, creating new collective works, for resale or redistribution to servers or lists, or reuse of any copyrighted component of this work in other works. Downloaded from http://hdl.handle.net/10072/67288 Griffith Research Online https://research-repository.griffith.edu.au A Tri-gram Based Feature Extraction Technique Using Linear Probabilities of Position Specific Scoring Matrix for Protein Fold Recognition Kuldip K. Paliwal, Member, IEEE, Alok Sharma, Member, IEEE, James Lyons, Abdollah Dhezangi, Member, IEEE protein structure in a reasonable amount of time. Abstract— In biological sciences, the deciphering of a three The prime objective of protein fold recognition is to find the dimensional structure of a protein sequence is considered to be an fold of a protein sequence. Assigning of protein fold to a important and challenging task. The identification of protein folds protein sequence is a transitional stage in the recognition of from primary protein sequences is an intermediate step in three dimensional structure of a protein. The protein fold discovering the three dimensional structure of a protein. This can be done by utilizing feature extraction technique to accurately recognition broadly covers feature extraction task and extract all the relevant information followed by employing a classification task. -

Structure, Function, and Mechanism of the Core Circadian Clock in Cyanobacteria
Structure, function, and mechanism of the core circadian clock in cyanobacteria Jeffrey A. Swan1, Susan S. Golden2,3, Andy LiWang3,4, Carrie L. Partch1,3* 1Chemistry and Biochemistry, University of California Santa Cruz, Santa Cruz, CA 95064 2Department of Molecular Biology, University of California San Diego, La Jolla, CA 92093 3Center for Circadian Biology and Division of Biological Sciences, University of California San Diego, La Jolla, CA 92093 4Chemistry and Chemical Biology, University of California Merced, Merced, CA 95343 *Correspondence to: [email protected] Running title: Structural review of the cyanobacterial clock Keywords: structural biology, circadian rhythm, circadian clock, cyanobacteria, protein dynamic, protein-protein interaction, post-translational modification, post-translational-oscillator, KaiABC Abbreviations: TTFL, transcription-translation feedback loop; PTO, post-translational oscillator; ATP, adenosine triphosphate; ADP, adenosine diphosphate; PsR, pseudo-receiver; HK, histidine kinase; RR, response regulator ______________________________________________________________________________ Abstract intertwined functions: timekeeping, entrainment Circadian rhythms enable cells and and output signaling. organisms to coordinate their physiology with the Timekeeping is achieved through a cycle of cyclic environmental changes that come as a result slow biochemical processes that together set the of Earth’s light/dark cycles. Cyanobacteria make period of oscillation close to 24 hours. For this use of a post-translational -

The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor
The thioredoxin encoded by the Rod-derived Cone Viability Factor gene protects cone photoreceptors against oxidative stress Xin Mei, Antoine Chaffiol, Christo Kole, Ying Yang, Géraldine Millet-Puel, Emmanuelle Clérin, Najate Aït-Ali, Jean Bennett, Deniz Dalkara, José-Alain Sahel, et al. To cite this version: Xin Mei, Antoine Chaffiol, Christo Kole, Ying Yang, Géraldine Millet-Puel, et al.. The thioredoxin encoded by the Rod-derived Cone Viability Factor gene protects cone photoreceptors against oxidative stress. Antioxidants and Redox Signaling, Mary Ann Liebert, 2016, 10.1089/ars.2015.6509. hal- 01297482 HAL Id: hal-01297482 https://hal.sorbonne-universite.fr/hal-01297482 Submitted on 4 Apr 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Original Research Communication The thioredoxin encoded by the Rod-derived Cone Viability Factor gene protects cone photoreceptors against oxidative stress Xin Mei1,2,3, Antoine Chaffiol1,2,3, Christo Kole1,2,3, Ying Yang1,2,3, Géraldine Millet-Puel1,2,3, Emmanuelle Clérin1,2,3, Najate Aït-Ali1,2,3, Jean Bennett4, Deniz Dalkara1,2,3, José-Alain Sahel1,2,3, Jens Duebel1,2,3 and Thierry Léveillard1,2,3 1 INSERM, U968, Paris, F-75012, France 2 Sorbonne Universités, UPMC Univ Paris 06, UMR_S 968, Institut de la Vision, Paris, F-75012, 3 CNRS, UMR_7210, Paris, F-75012, France 4 Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA, USA Corresponding author: Thierry Léveillard, Department of Genetics, Institut de la Vision, INSERM UMR-S 968, 17, rue Moreau, 75012 Paris, France. -

Small Molecule Diselenides As Probes of Oxidative Protein Folding
Research Collection Doctoral Thesis Small molecule diselenides as probes of oxidative protein folding Author(s): Beld, Joris Publication Date: 2009 Permanent Link: https://doi.org/10.3929/ethz-a-005950993 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library Diss. ETH No. 18510 Small molecule diselenides as probes of oxidative protein folding. A dissertation submitted to ETH Zürich For the degree of Doctor of Sciences Presented by Joris Beld MSc. University of Twente, Enschede Born February 15, 1978 Citizen of the Netherlands Accepted on the recommendation of Prof. Dr. Donald Hilvert, examiner Prof. Dr. Bernhard Jaun, co‐examiner Zürich 2009 How to work better. Do one thing at a time Know the problem Learn to listen Learn to ask questions Distinguish sense from nonsense Accept change as inevitable Admit mistakes Say it simple Be calm Smile Peter Fischli and David Weiss, 1991 To my family. Publications Parts of this thesis have been published. Beld, J., Woycechowsky, K.J., and Hilvert, D. Small molecule diselenides catalyze oxidative protein folding in vivo, submitted (2009) Beld, J., Woycechowsky, K.J., and Hilvert, D. Diselenide resins for oxidative protein folding, patent application EP09013216 (2009) Beld, J., Woycechowsky, K. J., and Hilvert, D. Catalysis of oxidative protein folding by small‐ molecule diselenides, Biochemistry 47, 6985‐6987 (2008) Beld, J., Woycechowsky, K.J., and Hilvert, D. Selenocysteine as a probe of oxidative protein folding, Oxidative folding of Proteins and Peptides, edited by J. -

Protein Folding and Assembly
CHEMICAL BIOLOGY / BIOLOGICAL CHEMISTRY 281 CHIMIA 2001, 55. No.4 Chimia 55 (2001) 281-285 © Schweizerische Chemische Gesellschaft ISSN 0009-4293 Protein Folding and Assembly Rudi Glockshuber* Abstract: One of the central dogmas in biochemistry is the view that the biologically active, three-dimensional structure of a protein is unique and exclusively determined by its amino acid sequence, and that the active conformation of a protein represents its state of lowest free energy in aqueous solution. Despite a large number of novel experiments supporting this view, including an exponentially increasing number of solved three- dimensional protein structures, it is still impossible to predict the tertiary structure of a protein from knowledge of its amino acid sequence alone. Towards the goal of identifying general principles underlying the mechanism of protein folding in vitro and in vivo, we are pursuing several projects that are briefly described in this article: (1) Circular permutation of proteins as a tool to study protein folding, (2) Catalysis of disulfide bond formation during protein folding, (3) Assembly of adhesive type 1 pili from Escherichia coli strains, and (4) Structure and folding of the mammalian prion protein. Keywords: Catalysis of disulfide bond formation' Prion proteins· Protein assembly' Protein folding' Type 1 pili During the last 20 years, the field of pro- molecular chaperones which assist the dom circular permutation analysis of a tein folding has gained enormous general protein folding process by preventing ir- protein using the periplasmic disulfide attention in the life sciences. First, this is reversible aggregation of unfolded and oxidoreductase DsbA from E. coli as a because large quantities of biologically partially folded polypeptide chains, and model system [2]. -
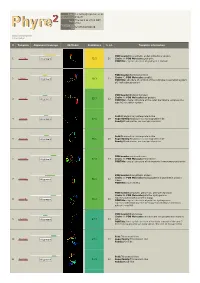
Hit Report.Pdf
Email [email protected] Description P18472 Thu Jan 5 11:37:00 GMT Date 2012 Unique Job 5279f5cb5050d57d ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:biosynthetic protein/structural protein 1 c2fu3A_ Alignment 72.2 20 Chain: A: PDB Molecule:gephyrin; PDBTitle: crystal structure of gephyrin e-domain PDB header:bacterial protein Chain: C: PDB Molecule:comb10; 2 c2bhvC_ 60.3 21 Alignment PDBTitle: structure of comb10 of the com type iv secretion system of2 helicobacter pylori PDB header:transport protein Chain: V: PDB Molecule:traf protein; 3 c3jqoV_ 57.7 22 Alignment PDBTitle: crystal structure of the outer membrane complex of a type iv2 secretion system Fold:Rhabdovirus nucleoprotein-like 4 d2gtta1 Alignment 57.5 29 Superfamily:Rhabdovirus nucleoprotein-like Family:Rhabdovirus nucleocapsid protein Fold:Rhabdovirus nucleoprotein-like 5 d2gica1 Alignment 55.1 16 Superfamily:Rhabdovirus nucleoprotein-like Family:Rhabdovirus nucleocapsid protein PDB header:oxidoreductase 6 c3emxB_ Alignment 52.0 17 Chain: B: PDB Molecule:thioredoxin; PDBTitle: crystal structure of thioredoxin from aeropyrum pernix PDB header:biosynthetic protein Chain: A: PDB Molecule:molybdopterin biosynthesis protein 7 c2nqqA_ 51.9 19 Alignment moea; PDBTitle: moea r137q PDB header:structural genomics, unknown function Chain: B: PDB Molecule:putative hydrogenase expression/formation protein hupg; 8 c2qsiB_ 50.2 20 Alignment PDBTitle: crystal structure of putative hydrogenase expression/formation -

The Homeobox Gene Chx10/Vsx2 Regulates Rdcvf Promoter Activity in the Inner Retina
THE HOMEOBOX GENE CHX10/VSX2 REGULATES RDCVF PROMOTER ACTIVITY IN THE INNER RETINA 1 2 1 1 Sacha ReichmanP ,P Ravi Kiran Reddy KalathurP ,P Sophie LambardP ,P Najate Ait-Ali , 3 2 2 2 1,3 Yanjiang Yang , Aurélie LardenoisP ,P Raymond RippP P Olivier PochP ,P Donald J. ZackP ,P 1 1 José-Alain SahelP P and Thierry Léveillard*P .P 1 Department of Genetics, Institut de la Vision, INSERM T Université Pierre et Marie Curie- 2 Paris6, UMR-S 968, Paris, F-75012 France,TP Institut de Génétique et de Biologie Moléculaire 3 et Cellulaire T (IGBMC),T 67404T Illkirch CEDEXT TFrance,P Wilmer Ophthalmological Institute, Johns Hopkins University School of Medicine, 600 N. Wolfe St. T Baltimore,T T MD 21287 USA. *Corresponding author: Thierry Léveillard, Department of Genetics, Institut de la Vision, INSERM, UPMC Univ Paris 06, UMR-S 968, CNRS 7210, Paris, F-75012 France. Tel : 33 1 53 46 25 48, Fax : 33 1 53 46 25 02, Email : [email protected] ABSTRACT Rod-derived Cone Viability Factor (RdCVF) is a trophic factor with therapeutic potential for the treatment of retinitis pigmentosa, a retinal disease that commonly results in blindness. RdCVF is encoded by Nucleoredoxin-like 1 (Nxnl1), a gene homologous with the family of thioredoxins that participate in the defense against oxidative stress. RdCVF expression is lost after rod degeneration in the first phase of retinitis pigmentosa, and this loss has been implicated in the more clinically significant secondary cone degeneration that often occurs. Here we describe a study of the Nxnl1 promoter using an approach that combines promoter and transcriptomic analysis. -
Target Highlights from the First Post‐
Received: 6 July 2017 | Revised: 19 September 2017 | Accepted: 25 September 2017 DOI: 10.1002/prot.25392 RESEARCH ARTICLE Target highlights from the first post-PSI CASP experiment (CASP12, May–August 2016) Andriy Kryshtafovych1 | Reinhard Albrecht2 | Arnaud Basle3 | Pedro Bule4 | Alessandro T. Caputo5 | Ana Luisa Carvalho6 | Kinlin L. Chao7 | Ron Diskin8 | Krzysztof Fidelis1 | Carlos M. G. A. Fontes4 | Folmer Fredslund9 | Harry J. Gilbert3 | Celia W. Goulding10 | Marcus D. Hartmann2 | Christopher S. Hayes11 | Osnat Herzberg7,12 | Johan C. Hill5 | Andrzej Joachimiak13,14 | Gert-Wieland Kohring15 | Roman I. Koning16,17 | Leila Lo Leggio9 | Marco Mangiagalli18 | Karolina Michalska13 | John Moult19 | Shabir Najmudin4 | Marco Nardini20 | Valentina Nardone20 | Didier Ndeh3 | Thanh-Hong Nguyen21 | Guido Pintacuda22 | Sandra Postel23 | Mark J. van Raaij21 | Pietro Roversi5,24 | Amir Shimon8 | Abhimanyu K. Singh25 | Eric J. Sundberg26 | Kaspars Tars27,28 | Nicole Zitzmann5 | Torsten Schwede29 1Genome Center, University of California, Davis, 451 Health Sciences Drive, Davis, California 95616 2Department of Protein Evolution, Max Planck Institute for Developmental Biology, Tubingen,€ 72076, Germany 3Institute for Cell and Molecular Biosciences, University of Newcastle, Newcastle upon Tyne NE2 4HH, United Kingdom 4CIISA - Faculdade de Medicina Veterinaria, Universidade de Lisboa, Avenida da Universidade Tecnica, 1300-477, Portugal, Lisboa 5Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1