Demographic and Prognostic Factors of Optic Nerve Astrocytoma
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

A Case of Intramedullary Spinal Cord Astrocytoma Associated with Neurofibromatosis Type 1
KISEP J Korean Neurosurg Soc 36 : 69-71, 2004 Case Report A Case of Intramedullary Spinal Cord Astrocytoma Associated with Neurofibromatosis Type 1 Jae Taek Hong, M.D.,1 Sang Won Lee, M.D.,1 Byung Chul Son, M.D.,1 Moon Chan Kim, M.D.2 Department of Neurosurgery,1 St. Vincent Hospital, The Catholic University of Korea, Suwon, Korea Department of Neurosurgery,2 Kangnam St. Mary's Hospital, The Catholic University of Korea, Seoul, Korea The authors report a symptomatic intramedullary spinal cord astrocytoma in the thoracolumbar area associated with neurofibromatosis type 1 (NF-1). A 38-year-old woman presented with paraparesis. Magnetic resonance imaging revealed an intramedullary lesion within the lower thoracic spinal cord and conus medullaris, which was removed surgically. Pathological investigation showed anaplastic astrocytoma. This case confirms that the diagnosis criteria set by the National Institute of Health Consensus Development Conference can be useful to differentiate ependymoma from astrocytoma when making a preoperative diagnosis of intramedullary spinal cord tumor in patients of NF-1. KEY WORDS : Astrocytoma·Intramedullary cord tumor·Neurofibromatosis. Introduction eurofibromatosis type 1 (NF-1), also known as von N Recklinghausen's disease, is one of the most common autosomal dominant inherited disorders with an incidence of 1 in 3,000 individuals and is characterized by a predisposition to tumors of the nervous system5,6,12,16). Central nervous system lesions associated with NF-1 include optic nerve glioma and low-grade gliomas of the hypothalamus, cerebellum and brain stem6,10). Since the introduction of magnetic resonance(MR) imaging, Fig. 1. Photograph of the patient's back shows multiple subcutaneous incidental lesions with uncertain pathological characteristic nodules (black arrow) and a cafe-au-lait spot (white arrow), which have been a frequent finding in the brain and spinal cord of are typical of NF-1. -

Pediatric Orbital Tumors and Lacrimal Drainage System
Pediatric Orbital Tumors and Lacrimal Drainage System Peter MacIntosh, MD University of Illinois • No financial disclosures Dermoid Cyst • Congenital • Keratinized epidermis • Dermal appendage • Trapped during embryogenesis • 6% of lesions • 40-50% of orbital pediatric orbital lesion • Usually discovered in the first year of life • Painless/firm/subQ mass • Rarely presents as an acute inflammatory lesion (Rupture?) • Frontozygomatic (70%) • Maxillofrontal (20%) suture Imaging - CT • Erosion/remodeling of bone • Adjacent bony changes: “smooth fossa” (85%) • Dumbell dermoid: extraorbital and intraorbital components through bony defect Imaging - MRI • Encapsulated • Enhancement of wall but not lumen Treatment Options • Observation • Risk of anesthesia • Surgical Removal • Changes to bone • Rupture of cyst can lead to acute inflammation • Irrigation • Abx • Steroids Dermoid INFANTILE/Capillary Hemangioma • Common BENIGN orbital lesion of children • F>M • Prematurity • Appears in 1st or 2nd week of life • Soft, bluish mass deep to the eyelid • Superonasal orbit • Rapidly expands over 6-12 months • Increases with valsalva (crying) • Clinical findings • Proptosis Astigmatism • Strabismus Amblyopia INFANTILE/Capillary Hemangioma • May enlarge for 1-2 years then regress • 70-80% resolve before age 7 • HIGH flow on doppler • Kasabach-Merritt Syndrome • Multiple large visceral capillary hemangiomas • Sequestration of platelets into tumor • Consumptive thrombocytopenia • Supportive therapy and treat underlying tumor • Complications • DIC • death •Homogenous -

Neuro-Oncology 1 XX(XX), 1–12, 2016 | Doi:10.1093/Neuonc/Now267
Neuro-Oncology 1 XX(XX), 1–12, 2016 | doi:10.1093/neuonc/now267 Defining the temporal course of murine neurofibromatosis-1 optic gliomagenesis reveals a therapeutic window to attenuate retinal dysfunction Joseph A. Toonen, Yu Ma, and David H. Gutmann Department of Neurology, Washington University School of Medicine (WUSM), St Louis, Missouri (J.A.T., Y.M., D.H.G.) Corresponding Author: David H. Gutmann, MD, PhD, Department of Neurology, Washington University, Box 8111, 660 S. Euclid Avenue, St. Louis MO 63110 ([email protected]). Abstract Background. Optic gliomas arising in the neurofibromatosis type 1 (NF1) cancer predisposition syndrome cause reduced visual acuity in 30%–50% of affected children. Since human specimens are rare, genetically engineered mouse (GEM) models have been successfully employed for preclinical therapeutic discovery and validation. However, the sequence of cellular and molecular events that culminate in retinal dysfunction and vision loss has not been fully defined relevant to potential neuroprotective treatment strategies. Methods. Nf1flox/mut GFAP-Cre (FMC) mice and age-matched Nf1flox/flox (FF) controls were euthanized at defined intervals from 2 weeks to 24 weeks of age. Optic nerve volumes were measured, and optic nerves/retinae analyzed by immunohistochemistry. Optical coherence tomography (OCT) was performed on anesthetized mice. FMC mice were treated with lovastatin from 12 to 16 weeks of age. Results. The earliest event in tumorigenesis was a persistent elevation in proliferation (4 wk), which preceded sustained microglia numbers and incremental increases in S100+ glial cells. Microglia activation, as evidenced by increased interleukin (IL)-1β expression and morphologic changes, coincided with axonal injury and retinal ganglion cell (RGC) apoptosis (6 wk). -

Clinical Manifestations of Hypothalamic Tumors*
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 10, No. 6 Copyright © 1980, Institute for Clinical Science, Inc. Clinical Manifestations of Hypothalamic Tumors* ADOLFO D. GARNICA, M.D., MICHAEL L. NETZLOFF, M.D.,f and A. L. ROSENBLOOM, M.D. Department of Pediatrics, University of Florida College of Medicine, Gainesville, FL 32610 and f Department of Human Development, Michigan State University East Lansing, MI 88823 ABSTRACT The regulatory function of the central nervous system encompasses di verse endocrine, metabolic, and behavioral processes. Many of these origi nate, are integrated, or are coordinated through hypothalamic pathways or nuclei. Thus, tumors affecting areas projecting to the hypothalamus, tumors of the hypothalamus, and tumors invading or compressing the hypothalamus can produce abnormalities of hypothalamic function. Introduction tary.4,7,31 A secretory function for certain hypothalamic neurons was postulated in Until recently, no endocrine disorder 1928 and subsequently confirmed by the directly attributable to hypothalamic dys demonstration of hormone synthesis in function had been recognized, and the the supraoptic and paraventricular nu majority of endocrine-metabolic homeo clei.28,53 Moreover, observations on the static processes were acknowledged to be effects of environment on the menstrual under the control of the anterior pitui cycles of women and the study of repro tary.48,49 However, in 1901 Frohlich re ductive cycles in animals have shown a ported a patient with a suprasellar tumor, functional connection -

Low-Grade Central Nervous System Tumors
Neurosurg Focus 12 (2):Article 1, 2002, Click here to return to Table of Contents Low-grade central nervous system tumors M. BEATRIZ S. LOPES, M.D., AND EDWARD R. LAWS, JR., M.D. Departments of Pathology (Neuropathology) and Neurological Surgery, University of Virginia Health Sciences Center, Charlottesville, Virginia Low-grade tumors of the central nervous system constitute 15 to 35% of primary brain tumors. Although this cate- gory of tumors encompasses a number of different well-characterized entities, low-grade tumors constitute every tumor not obviously malignant at initial diagnosis. In this brief review, the authors discuss the pathological classification, diagnostic procedures, treatment, and possible pathogenic mechanisms of these tumors. Emphasis is given in the neu- roradiological and pathological features of the several entities. KEY WORDS • glioma • astrocytoma • treatment outcome Low-grade gliomas of the brain represent a large pro- toses. The pilocytic (juvenile) astrocytoma is a character- portion of primary brain tumors, ranging from 15 to 35% istic, more circumscribed lesion occurring primarily in in most reported series.1–5 They include a remarkable di- childhood and with a predilection for being located in the versity of lesions, all of which have been lumped together cerebellum. It usually appears as a cystic tumor with a under the heading of "low-grade glioma." This category mural nodule. The tumor tissue itself may have features of includes virtually every tumor of glial origin that is not microcystic degeneration and Rosenthal fibers which are overtly malignant at the time of initial diagnosis. degenerative structures in the astrocytic processes. Other reasonably common types of low-grade gliomas include CLASSIFICATION OF GLIOMAS the low-grade oligodendroglioma and the low-grade ependymoma, which is usually anatomically related to the Table 1 provides a classification of low-grade tumors of ventricular ependymal lining. -

Cranial Nerve Disorders: Clinical Manifestations and Topographyଝ
Radiología. 2019;61(2):99---123 www.elsevier.es/rx UPDATE IN RADIOLOGY Cranial nerve disorders: Clinical manifestations and topographyଝ a,∗ a b c M. Jorquera Moya , S. Merino Menéndez , J. Porta Etessam , J. Escribano Vera , a M. Yus Fuertes a Sección de Neurorradiología, Hospital Clínico San Carlos, Madrid, Spain b Servicio de Neurología, Hospital Clínico San Carlos, Madrid, Spain c Neurorradiología, Hospital Ruber Internacional, Madrid, Spain Received 17 November 2017; accepted 27 September 2018 KEYWORDS Abstract The detection of pathological conditions related to the twelve cranial pairs rep- Cranial pairs; resents a significant challenge for both clinicians and radiologists; imaging techniques are Cranial nerves; fundamental for the management of many patients with these conditions. In addition to knowl- Cranial neuropathies; edge about the anatomy and pathological entities that can potentially affect the cranial pairs, Neuralgia; the imaging evaluation of patients with possible cranial pair disorders requires specific exami- Cranial nerve palsy nation protocols, acquisition techniques, and image processing. This article provides a review of the most common symptoms and syndromes related with the cranial pairs that might require imaging tests, together with a brief overview of the anatomy, the most common underlying processes, and the most appropriate imaging tests for different indications. © 2018 SERAM. Published by Elsevier Espana,˜ S.L.U. All rights reserved. PALABRAS CLAVE Sintomatología derivada de los pares craneales: Clínica y topografía Pares craneales; Resumen La detección de la patología relacionada con los doce pares craneales representa Nervios craneales; un importante desafío, tanto para los clínicos como para los radiólogos. Las técnicas de imagen Neuropatía de pares craneales; son fundamentales para el manejo de muchos de los pacientes. -

Immunohistochemical Properties of Human Optic Nerve Glioma
Investigative Ophthalmology & Visual Science, Vol. 32, No. 9, August 1991 Copyright © Association for Research in Vision and Ophthalmology Immunohistochemical Properties of Human Optic Nerve Glioma Evidence of Type 1 Astrocyte Origin Paul E. Cutarelli,* Uros R. Roessmann,* Robert H. Miller, f Charles S. Spechr4 and Hans E. Grossniklaus§ The peroxidase-antiperoxidase method was used to study ten surgically obtained human optic nerve gliomas (pilocytic astrocytomas). All tissues were formalin fixed and paraffin embedded. Primary antisera included glial fibrillary acidic protein (GFAP), HNK-1 (type 1 astrocyte precursor marker), A2B5 (type 2 astrocyte precursor marker), S-100, vimentin, myelin basic protein (MBP), laminin, keratin, cytokeratin, epithelial membrane antigen (EMA), and neuron-specific enolase (NSE). Neo- plastic astrocytes in optic nerve gliomas stained with GFAP, HNK-1, S-100, and vimentin. Oligoden- drocytes and myelin sheaths stained for MBP, and NSE stained surviving axons in the tumors. Neo- plastic astrocytes did not stain for A2B5, keratin, cytokeratin, EMA, or laminin. These results suggest that human optic nerve gliomas (pilocytic astrocytomas) arise from type 1 astrocytes. Invest Ophthal- mol Vis Sci 32:2521-2524,1991 As a result of the variable histopathologic appear- with the identification in the rat central nervous sys- ance of optic nerve glioma, several classifications of tem of a macroglial progenitor cell capable of forming the origin of the proliferating astrocyte comprising astrocytes and oligodendrocytes.4-5 To characterize these tumors have arisen.1"3 The light microscopic ap- more fully the cellular elements present in optic nerve pearance of certain cells in optic nerve glioma encour- glioma and to identify the origin of the proliferating aged some authors to interpret these tumors as oligo- astrocyte, we immunostained ten human optic nerve dendrogliomas.1 The detection of neurofibrils in these gliomas. -
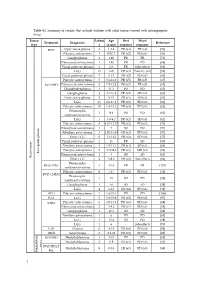
Table S1. Summary of Studies That Include Children with Solid Tumors Treated with Antiangiogenic Drugs
Table S1. Summary of studies that include children with solid tumors treated with antiangiogenic drugs. Tumor Patient Age Best Worst Treatment Diagnostic Reference type s (years) response response BVZ Optic nerve glioma 4 1.2-4 PR (x4) PR (x4) [56] Pilocytic astrocytoma 3 0.92-7 PR (x2) PD (x1) [56] Ganglioglioma 1 1.92 PR PR [56] Pilomyxoid astrocytoma 1 1.92 PR PD [68] Visual pathway glioma 1 2.6 PR Side effects [66] LGG 15 1-20 CR (x3) Toxicity (x1) [58] Visual pathway glioma 3 6-13 PR (x2) PD (x1) [60] Pilocytic astrocytoma 5 3.1-11.2 PR (x5) PR (x5) [65] BVZ+IRO Pilomyxoid astrocytoma 2 7.9-12.2 PR (x2) PR (x2) [65] Oligodendroglioma 1 11.1 PD PD [65] Ganglioglioma 3 4.1-16.8 PR (x2) SD (x1) [65] Optic nerve glioma 2 9-15 PR (x1) SD (x1) [63] LGG 35 0.6-17.6 PR (x2) PD (x8) [61] Pilocytic astrocytoma 10 1.8-15.3 PR (x4) PD (x1) [62] Pleomorphic 1 9.4 PD PD [62] xanthoastrocytoma LGG 5 3.9-9.2 PR (x2) SD (x3) [62] Pilocytic astrocytoma 4 4.67-12.17 PR (x2) PD (x3) [57] Pilomyxoid astrocytoma 1 3 SD PD [57] Fibrillary astrocytoma 3 1.92-11.08 CR (x1) PD (x3) [57] Other LGG 6 1-13.42 PR (x2) PD (x6) [57] Visual pathway glioma 1 11 PR SD [60] Fibrillary astrocytoma 3 1.5-11.1 CR (x1) SD (x1) [64] Pilocytic astrocytoma 2 3.75-9.8 PR (x1) MR (x1) [64] Low-grade glioma Pilomyxoid astrocytoma 1 3 SD SD [64] Brain tumor Brain tumor Other LGG 4 3-9.6 PR (x2) Side effects [64] Pleomorphic BVZ+TMZ 1 13.4 PR PR [153] xanthoastrocytoma Pilocytic astrocytoma 9 5-17 PR (x3) PD (x3) [59] BVZ+CHEM Pleomorphic 1 18 SD PD [59] xanthoastrocytoma Ganglioglioma -

Differential Diagnosis of Sellar Masses
~~ ~~ ~ ADVANCES IN PITUITARY TUMOR THERAPY 0889-8529/99 $8.00 + .OO DIFFERENTIAL DIAGNOSIS OF SELLAR MASSES Pamela U. Freda, MD, and Kalmon D. Post, MD Pituitary adenomas are the most common cause of a mass in the sella. In as many as 9% of cases, other etiologies are responsible for mass lesions in the sellar regions4,13' (Table 1). The differential diagnosis of nonpituitary sellar masses is broad and includes cell rest tumors, germ cell tumors, gliomas, menin- giomas, metastatic tumors, vascular lesions, and granulomatous, infectious, and inflammatory processes (Table 2). Differentiating among these potential etiolo- gies may not always be straightforward because many of these lesions, tumorous and nontumorous, may mimic the clinical, endocrinologic, and radiographic presentations of pituitary adenomas. In some cases, there are no features that clearly distinguish the unusual etiologies from the clinically nonfunctioning pituitary adenoma. In others, certain endocrine, neurologic, and radiographic findings that are more characteristic of patients with a nonpituitary sellar mass may be present and can help in their differentiation. Correct preoperative diag- nosis is clinically important because the treatment of choice for many of these nonpituitary sellar masses differs from that of a pituitary tumor. This article provides an overview of the clinical and radiographic characteristics of both pituitary tumors and the nonpituitary lesions found in the sellar/parasellar region and discusses in detail the specific nonpituitary etiologies of the sellar mass. SIGNS AND SYMPTOMS OF PITUITARY TUMORS Pituitary tumors vary in presentation. Clinical findings depend largely on whether the tumor is hormone secreting or clinically nonfunctioning, on the size and pattern of tumor growth, and on whether normal pituitary gland function is disrupted. -

Optic Nerve: Developmental Anomalies and Common Tumors
DOI: 10.5772/intechopen.80326 ProvisionalChapter chapter 3 Optic Nerve: Developmental Anomalies and Common Tumors HindHind Alkatan, Alkatan, Daniah AlshowaeirDaniah Alshowaeir and TariqTariq Alzahem Alzahem Additional information is available at the end of the chapter http://dx.doi.org/10.5772/intechopen.80326 Abstract The optic nerve, also known as the second cranial nerve, is composed of axons that trans- mit visual information from the neurosensory retina to the visual cortex. There are mul- tiple pathologies that can affect the human optic nerve. Congenital anomalies of the optic nerve include myelinated nerve fibers, morning glory syndrome, optic nerve choristoma, optic nerve coloboma, optic nerve hypoplasia and aplasia, and others. Tumors that can affect the optic nerve (ON) may occur primarily from within the nerve itself, from the sur- rounding optic nerve sheath (ONS), or secondarily spreading to the nerve from a distant site. They include optic pathway glioma, medulloepithelioma, oligodendroglioma, optic nerve sheath meningioma, and others. Here in this chapter, we will review the optic nerve anatomy, embryology, and physiology in addition to assessment of optic nerve function. Moreover, the clinical features, imaging findings, pathology, and treatment options of the most common and some rare congenital anomalies and primary tumors of the ON and sheath will be reviewed. Keywords: myelinated nerve fibers, morning glory syndrome, optic nerve choristoma, optic nerve coloboma, optic nerve hypoplasia, aplasia, optic nerve tumor, glioma, meningioma, ganglioglioma, medulloepithelioma, hemangioblastoma, oligodendroglioma 1. Introduction Visual perception occurs when light stimulus in the surrounding environment converts to nerve impulses at the level of photoreceptors, which then reach the brain to be processed. -

CT-Scan Findings of Orbital Mass Among Pediatric Patients at a Tertiary Care Hospital in Bangladesh
ORIGINAL ARTICLE CT-Scan Findings of Orbital Mass Among Pediatric Patients at a Tertiary Care Hospital in Bangladesh *M A Rahman1, SA Azad2, N Islam3, S Amin4, MZ Haque5, MHM Kamal6, Mukhdira7 1*Dr Meher Angez Rahman. Consultant, Department of Radiology and Imaging Anwar Khan Modern Medical College and Hospital 2Prof Salauddin Al Azad, Professor, Department of Radiology & Imaging, BSMMU 3Dr. Nazrul Islam, Associate Professor, Department of Radiology & Imaging, BSMMU 4Dr. Prof Sadrul Amin, Associate Professor, Department of Radiology & Imaging, BSMMU 5Dr. Md ziaul Haque, ex-Associate Professor, Department of Radiology & Imaging, BSMMU 6Dr. Mahmud Hasan Mostofa Kamal, ex-Assistant Professor, Department of Radiology & Imaging, BSMMU 7Dr. Mukhdira, Lecturer, Department of Physiology, Community Based Medical College *Corresponding Author Date of submission: 08.05.2016 Date of acceptance:12.11.2016 ABSTRACT Background: CT-Scan for the detection of orbital mass among pediatric patients is very important noninvasive radiological modality. The purpose of the study was to find out CT-Scan findings of orbital mass among pediatric patients in a tertiary care hospital. Methodology: This is a cross sectional study was carried out in Ophthalmology and Radiology and Imaging department of National Institute of Ophthalmology (NIO) from January 2012 to December 2013. All the patient below 18 years of age presented with suspected orbital mass at Ophthalmology and Radiology and Imaging department of NIO and performed CT- Scan of orbit for diagnosis of the disease and also done histopathology after operation was enrolled in this study. Results: In this study it was observed that a total of 29 cases identified as malignant evaluated by CT, among them 27 cases were true positive and 2 cases were false positive. -

Orbital Mass in a Child
Orbital Mass in a Child Edward G. Buckley, MD Banks Anderson Professor of Ophthalmology Duke University Orbital Tumors/Lesions in Children • Malformations • Choristomas - Hemangioma, Lymphangioma, Varix, Neurofibroma • Primary Neoplasms - Glioma, Rhabdomyosarcoma, fibrous dysplasia • Secondary tumors – Astrocytoma, medulloepithelioma • Metastatic tumors – Neuroblastoma, Wilm’s , Ewing’s sarcoma • Leukemias / Lymphoma – Burkitt’s • Histiocytoses/Xanthogranuloma- esosinophilic granuloma • Inflammations- Pseudotumor, myositis • Infections – orbital cellulitis How to evaluate a child with orbital mass? Neuro-imaging ! Which neuro-imaging test is best ? CT scans are superior in most cases MRI may be desirable in certain cases when optic nerve dysfunction is present How to evaluate an orbital mass in a child? Ways to: How to determine: Classify ? Seriousness ? Categorize ? Urgency ? Compartmentalize ? Morbidity ? Is it Rapidly Expanding ?….. • Cellulitis/abscess • Pseudotumor/myositis • Hemangioma • Rhabdomyosarcoma • Neuroblastoma • Lymphoma • Eosinophilic granuloma Bilateral ?….. • Optic nerve glioma • Neuroblastoma • Leukemia • Lymphoma • Pseudotumor/myositis • Eosinophilic granuloma Eyelid echymosis ?…. • Neuroblastoma • Ewing’s sarcoma • Leukemia • Eosinophilic granuloma • Lymphanigoma Present at birth ?...... Microphthalmos with cyst Varix Teratoma Optic nerve glioma Capillary hemangioma Retinoblastoma Lymphangioma Neuroblastoma Dermoid cyst Neurofibroma Meningoencephalocele Juvrnile xanthogranuloma Intermittent ?.... Lympangioma Dermoid