Speciation Genomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Directory of Important Bird Areas in Vietnam KEY SITES for CONSERVATION
BirdLife International in Indochina and the Institute of Ecology and Biological Resources with financial support from Danida Directory of Important Bird Areas in Vietnam KEY SITES FOR CONSERVATION Andrew W. Tordoff (editor) with contributions from Dr Nguyen Cu, Jonathan C. Eames, Neil M. Furey, Le Manh Hung, Ha Quy Quynh, Adam M. Seward, Le Trong Trai, Nguyen Duc Tu and Dr Corinthe T. Zekveld This publication is a technical output of the Danida-funded project Improved conservation planning through institutional strengthening in Cambodia, Laos and Vietnam. Hanoi, November 2002 Project funder: Danida Editor: Andrew W. Tordoff Contributors: Dr Nguyen Cu Jonathan C. Eames Neil M. Furey Le Manh Hung Ha Quy Quynh Adam M. Seward Le Trong Trai Nguyen Duc Tu Dr Corinthe T. Zekveld Maps: Ha Quy Quynh Cover illustrations: Background: Forest canopy at Nam Cat Tien IBA by Andrew Tordoff. Left (top to bottom): Crocodile Lake at Nam Cat Tien IBA by Jonathan Eames; Sapria himalayana by Jonathan Eames; Delacour’s Langur Trachypithecus delacouri by Andrew Tordoff. Centre (top to bottom): Hornbill mobile by Andrew Tordoff; Mountain Scops Owl Otus spilocephalus by Jonathan Eames; Wreathed Hornbill Aceros undulatus by Jonathan Eames; Montane evergreen forest at Kon Ka Kinh IBA by Ben Hayes; Common Kingfisher Alcedo atthis by Ben Hayes. Right (top to bottom): Long-tailed Broadbill Psarisomus dalhousiae by Ben Hayes; Dragonfly by Ben Hayes; Riverine forest at Kon Cha Rang IBA by Ben Hayes; Caterpillars by Ben Hayes. Suggested citation: Tordoff, A. W. ed. (2002) Directory of Important Bird Areas in Vietnam: key sites for conservation. Hanoi: BirdLife International in Indochina and the Institute of Ecology and Biological Resources. -

ARK 2 Five Physically Haiidicapped Magpies Are Destroyed (Unless They Are Cunning and Hide in a Shed Until the Season Is Over)
Between us.... THE SONG_________ fl. V, £,.X R...A..L_I_A There is a land, where summer skies Are gleaming with a thousand dyes, Blending in witching harmonies; And grassy knoll and forest height Are flashing in the rosy light, And all above is azure bright - Australia! There is a land where honey flows Where laughing corn luxuriant grows, Land of the myrtle and the rose. On hill and plain the clustering vine Is gushing out with purple wine, And cups are quaffed to thee and thine - Australia! There is a land where treasures shine Deep in the dark unfathod'd mine, i?or worshippers at , Mammon's shrine; Where gold lies hid and rubies gleam, And fabled wealth no more doth seem The idle fancy of a dream - Aus tralia! There is a land where homesteads peep, From sunny plain and woodland steep, And love and joy bright vigils keep; Where the glad voice of childish glee Is mingled with the melody Of nature's hidden minstrelsy - Australia! There is a land where, floating free From mountain top to girdling sea, A proud flag waves exultingly; And freedom's song the banner bear - No shackled slave can breathe the air: Fairest of Britain's daughters fair - Australia! C.J. Carleton It comes, once an issue, a time to sit down and act ually do something creative yourself - it’s all very well to put two ARK two out a magazine written by others (l mean to say, they write better than you do), drawings by talented people, and the setti ng-out layed dox^n by your dear husband, xdio, after all, has had ten years in fanzine pubbing to decide what looks best layout. -

SICHUAN (Including Northern Yunnan)
Temminck’s Tragopan (all photos by Dave Farrow unless indicated otherwise) SICHUAN (Including Northern Yunnan) 16/19 MAY – 7 JUNE 2018 LEADER: DAVE FARROW The Birdquest tour to Sichuan this year was a great success, with a slightly altered itinerary to usual due to the closure of Jiuzhaigou, and we enjoyed a very smooth and enjoyable trip around the spectacular and endemic-rich mountain and plateau landscapes of this striking province. Gamebirds featured strongly with 14 species seen, the highlights of them including a male Temminck’s Tragopan grazing in the gloom, Chinese Monal trotting across high pastures, White Eared and Blue Eared Pheasants, Lady Amherst’s and Golden Pheasants, Chinese Grouse and Tibetan Partridge. Next were the Parrotbills, with Three-toed, Great and Golden, Grey-hooded and Fulvous charming us, Laughingthrushes included Red-winged, Buffy, Barred, Snowy-cheeked and Plain, we saw more Leaf Warblers than we knew what to do with, and marvelled at the gorgeous colours of Sharpe’s, Pink-rumped, Vinaceous, Three-banded and Red-fronted Rosefinches, the exciting Przevalski’s Finch, the red pulse of Firethroats plus the unreal blue of Grandala. Our bird of the trip? Well, there was that Red Panda that we watched for ages! 1 BirdQuest Tour Report: Sichuan Including Northern Yunnan 2018 www.birdquest-tours.com Our tour began with a short extension in Yunnan, based in Lijiang city, with the purpose of finding some of the local specialities including the rare White-speckled Laughingthrush, which survives here in small numbers. Once our small group had arrived in the bustling city of Lijiang we began our birding in an area of hills that had clearly been totally cleared of forest in the fairly recent past, with a few trees standing above the hillsides of scrub. -

Trip Report 17Th August to 3Rd September 2013
Sulawesi & Halmahera Wallacean Endemics Trip Report 17th August to 3rd September 2013 Lilac Kingfisher by David Hoddinott RBT Sulawesi & Halmahera 2013 Trip Report 2 Trip report compiled by Tour Leader: David Hoddinott Top 10 birds as voted by participants: 1. Standardwing 6. Azure Dollarbird 2. Maleo 7. Moluccan Owlet-nightjar 3. Sulawesi Masked Owl 8. Lilac Kingfisher 4. Ivory-breasted Pitta 9. Red-backed Thrush 5. Mountain Serin 10. Sulawesi Dwarf Kingfisher Tour Summary Surrounded to the north by the Philippines, to the east by New Guinea, the south by Australia and the west by Borneo, the two larger islands of Sulawesi and Halmahera form a significant part of central Indonesia’s nearly 15000 islands. We recorded over 100 endemics of a total trip list of 254 species, thus emphasising that this is certainly one of the endemic hotspots of the world! Our tour started off with an early morning visit to the limestone crags of Karaenta Forest, Sulawesi. Departing Makassar, we set off early to maximise our limited time as we had a flight to catch in the early afternoon. Arriving just as the sun’s first rays hit the treetops, we were soon enjoying wonderful sightings of stunning Grey-sided and Yellow-sided Flowerpeckers. You could actually feel the sense of excitement in the air as we notched up our first endemics. After enjoying our tea and coffee, we then quickly picked up our main target, the rather localised endemic Black-ringed Whit-eye, which showed splendidly as it sat just in front of us gobbling down some ripe fruit. -

GRUNDSTEN Sulawesi 0708
Birding South and Central Sulawesi (M. Grundsten, Sweden) 2016 South and Central Sulawesi, July 28th - August 5th 2016 Front cover Forest-dwelling Dwarf Sparrowhawk, Accipiter nanus, Anaso track, Lore Lindu NP (MG). Participants Måns Grundsten [email protected] (compiler and photos (MG)) Mathias Bergström Jonas Nordin, all Stockholm, Sweden. Highlights • Luckily escaping the previous extensive occlusion of Anaso track due to terrorist actions. Anaso track opened up during our staying. • A fruit-eating Tonkean Macaque at Lore Lindu. • Great views of hunting Eastern Grass Owls over paddies around Wuasa on three different evenings. • Seeing a canopy-perched Sombre Pigeon above the pass at Anaso track. • Flocks of Malias and a few Sulawesi Thrushes. • No less than three different Blue-faced Parrotfinches. • Purple-bearded Bee-eaters along Anaso track. • Finding Javan Plover at Palu salt pans, to our knowledge a significant range extension, previously known from Sulawesi mainly in Makassar-area. • Sulawesi Hornbill at Paneki valley. • Sulawesi Streaked Flycatcher at Paneki valley, a possibly new location for this recently described species. • Two days at Gunung Lompobattang in the south: Super-endemic Lompobattang Flycatcher, Black-ringed White-eye, and not-so-easy diminutive Pygmy Hanging Parrot. Logistics With limited time available this was a dedicated trip to Central and Southern Sulawesi. Jonas and Mathias had the opportunity to extend the trip for another week in the North while I had to return home. The trip was arranged with help from Nurlin at Palu-based Malia Tours ([email protected]). Originally we had planned to have a full board trip to Lore Lindu and also include seldom-visited Saluki in the remote western parts of Lore Lindu, a lower altitude part of Lore Lindu where Maleo occurs. -

The Avifauna of Lambusango Forest Reserve, Buton Island, South-East Sulawesi, with Additional Sightings from Southern Buton
FORKTAIL 28 (2012): 107–112 The avifauna of Lambusango Forest Reserve, Buton Island, south-east Sulawesi, with additional sightings from southern Buton T. E. MARTIN, D. J. KELLY, N. T. KEOGH, D. HERIYADI, H. A. SINGER & G. A. BLACKBURN Lambusango Forest Reserve occupies a large area of south-central Buton, the largest attendant island of Sulawesi, Indonesia. Buton is located off Sulawesi’s south-eastern peninsula and remains poorly known ornithologically. Bird surveys were undertaken in the reserve over eight eight-week long research seasons between June and August in 1999, 2001–2003, 2005, and 2008–2010. Variable radius circular- plot point counts were the primary census method, conducted as part of a long-term biodiversity monitoring programme in the reserve, although data were also collected from 840 mist-netting hours and approximately 2,560 hours of observational data. In total, 79 species were detected in the reserve, including 37 regional endemics (46.8% of the total avifaunal community) and four species considered by the IUCN to be globally threatened or Near Threatened. Additionally, a further 60 species (including two more Near Threatened species) were recorded in various habitats around southern Buton that were not detected in Lambusango Reserve, giving a total of 139 species records for the island. We believe that 51 of these species represent previously unpublished records for Buton. We present here a full account of our records from Lambusango Reserve and southern Buton, with additional details provided for threatened and Near Threatened species and new records of endemics. INTRODUCTION Lambusango Forest Reserve (5°10’–5°24’S 122°43’–123°07’E) is a 65,000 ha expanse of uninhabited tropical monsoon forest, Buton (formerly referred to as Butung) is the largest of Sulawesi’s encompassing much of south-central Buton. -
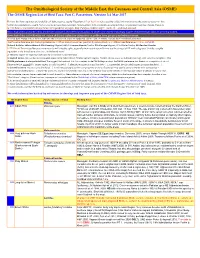
OSME List V3.4 Passerines-2
The Ornithological Society of the Middle East, the Caucasus and Central Asia (OSME) The OSME Region List of Bird Taxa: Part C, Passerines. Version 3.4 Mar 2017 For taxa that have unproven and probably unlikely presence, see the Hypothetical List. Red font indicates either added information since the previous version or that further documentation is sought. Not all synonyms have been examined. Serial numbers (SN) are merely an administrative conveninence and may change. Please do not cite them as row numbers in any formal correspondence or papers. Key: Compass cardinals (eg N = north, SE = southeast) are used. Rows shaded thus and with yellow text denote summaries of problem taxon groups in which some closely-related taxa may be of indeterminate status or are being studied. Rows shaded thus and with white text contain additional explanatory information on problem taxon groups as and when necessary. A broad dark orange line, as below, indicates the last taxon in a new or suggested species split, or where sspp are best considered separately. The Passerine Reference List (including References for Hypothetical passerines [see Part E] and explanations of Abbreviated References) follows at Part D. Notes↓ & Status abbreviations→ BM=Breeding Migrant, SB/SV=Summer Breeder/Visitor, PM=Passage Migrant, WV=Winter Visitor, RB=Resident Breeder 1. PT=Parent Taxon (used because many records will antedate splits, especially from recent research) – we use the concept of PT with a degree of latitude, roughly equivalent to the formal term sensu lato , ‘in the broad sense’. 2. The term 'report' or ‘reported’ indicates the occurrence is unconfirmed. -
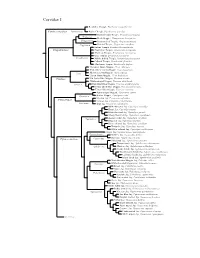
Corvidae Species Tree
Corvidae I Red-billed Chough, Pyrrhocorax pyrrhocorax Pyrrhocoracinae =Pyrrhocorax Alpine Chough, Pyrrhocorax graculus Ratchet-tailed Treepie, Temnurus temnurus Temnurus Black Magpie, Platysmurus leucopterus Platysmurus Racket-tailed Treepie, Crypsirina temia Crypsirina Hooded Treepie, Crypsirina cucullata Rufous Treepie, Dendrocitta vagabunda Crypsirininae ?Sumatran Treepie, Dendrocitta occipitalis ?Bornean Treepie, Dendrocitta cinerascens Gray Treepie, Dendrocitta formosae Dendrocitta ?White-bellied Treepie, Dendrocitta leucogastra Collared Treepie, Dendrocitta frontalis ?Andaman Treepie, Dendrocitta bayleii ?Common Green-Magpie, Cissa chinensis ?Indochinese Green-Magpie, Cissa hypoleuca Cissa ?Bornean Green-Magpie, Cissa jefferyi ?Javan Green-Magpie, Cissa thalassina Cissinae ?Sri Lanka Blue-Magpie, Urocissa ornata ?White-winged Magpie, Urocissa whiteheadi Urocissa Red-billed Blue-Magpie, Urocissa erythroryncha Yellow-billed Blue-Magpie, Urocissa flavirostris Taiwan Blue-Magpie, Urocissa caerulea Azure-winged Magpie, Cyanopica cyanus Cyanopica Iberian Magpie, Cyanopica cooki Siberian Jay, Perisoreus infaustus Perisoreinae Sichuan Jay, Perisoreus internigrans Perisoreus Gray Jay, Perisoreus canadensis White-throated Jay, Cyanolyca mirabilis Dwarf Jay, Cyanolyca nanus Black-throated Jay, Cyanolyca pumilo Silvery-throated Jay, Cyanolyca argentigula Cyanolyca Azure-hooded Jay, Cyanolyca cucullata Beautiful Jay, Cyanolyca pulchra Black-collared Jay, Cyanolyca armillata Turquoise Jay, Cyanolyca turcosa White-collared Jay, Cyanolyca viridicyanus -

Ultimate Sulawesi & Halmahera 2016
Minahassa Masked Owl (Craig Robson) ULTIMATE SULAWESI & HALMAHERA 4 - 24 SEPTEMBER 2016 LEADER: CRAIG ROBSON The latest Birdquest tour to Sulawesi and Halmahera proved to be another great adventure, with some stunning avian highlights, not least the amazing Minahassa Masked Owl that we had such brilliant views of at Tangkoko. Some of the more memorable highlights amongst our huge trip total of 292 species were: 15 species of kingfisher (including Green-backed, Lilac, Great-billed, Scaly-breasted, Sombre, both Sulawesi and Moluccan Dwarf, and Azure), 15 species of nightbird seen (including Sulawesi Masked and Barking Owls, Ochre-bellied and Cinnabar Boobooks, Sulawesi and Satanic Nightjars, and Moluccan Owlet-Nightjar), the incredible Maleo, Moluccan Megapode at point-blank range, Pygmy Eagle, Sulawesi, Spot-tailed and 1 BirdQuest Tour Report: Ultimate Sulawesi & Halmahera 2016 www.birdquest-tours.com Moluccan Goshawks, Red-backed Buttonquail, Great and White-faced Cuckoo-Doves, Red-eared, Scarlet- breasted and Oberholser’s Fruit Doves, Grey-headed Imperial Pigeon, Moluccan Cuckoo, Purple-winged Roller, Azure (or Purple) Dollarbird, the peerless Purple-bearded Bee-eater, Knobbed Hornbill, White Cockatoo, Moluccan King and Pygmy Hanging Parrots, Chattering Lory, Ivory-breasted, Moluccan and Sulawesi Pittas (the latter two split from Red-bellied), White-naped and Shining Monarchs, Maroon-backed Whistler, Piping Crow, lekking Standardwings, Hylocitrea, Malia, Sulawesi and White-necked Mynas, Red- backed and Sulawesi Thrushes, Sulawesi Streaked Flycatcher, the demure Matinan Flycatcher, Great Shortwing, and Mountain Serin. Moluccan Megapode, taking a break from all that digging! (Craig Robson) This year’s tour began in Makassar in south-west Sulawesi. Early on our first morning we drove out of town to the nearby limestone hills of Karaenta Forest. -

Eastern China
The magnificent Reeves's Pheasant was one of the many specialties seen on this tour (Brendan Ryan). EASTERN CHINA 3 – 27 MAY 2017 LEADER: HANNU JÄNNES Birdquest’s Eastern China tour, an epic 25 day journey across much of eastern China, focusses on an array of rare Chinese endemics and migrants, and this year’s tour once again proved a great success. The focus of the first part of the tour is to achieve good views of rarities like Spoon-billed Sandpiper, the critically endangered Blue-crowned (Courtois’s) Laughingthrush, the superb Cabot’s Tragopan and Elliot’s Pheasant and the ultra-rare Chinese Crested Tern. This was successfully achieved alongside a plethora of other much sought after species including White-faced Plover, Great Knot, stunning Saunders’s Gulls, Reed Parrotbill, eastern migrants, including Pechora Pipit, Japanese Robin, Japanese Paradise, Yellow-rumped, Narcissus and Mugimaki Flycatchers, and forest species like Brown-chested Jungle Flycatcher, White-necklaced Partridge, Silver Pheasant, Buffy and Moustached Laughingthrushes, Short-tailed Parrotbill, Fork-tailed Sunbird and the delightful Pied Falconet. Quite a haul! 1 BirdQuest Tour Report: Eastern China 2017 www.birdquest-tours.com Crested Ibis at Dongzhai Nature Reserve (Brendan Ryan). The second part of the tour, the ‘Northeast Extension’, visited a series of sites for various other Chinese specialities. Beginning in Wuhan, we bagged the amazing Reeves’s Pheasant and Crested Ibis, as well as stunners that included Fairy Pitta and Chestnut-winged Cuckoo. We then moved on to Jiaocheng for the fabulous Brown Eared Pheasants before flying on to Beijing, where the mountains of the nearby Hebei province yielded the endemic Chinese Beautiful Rosefinch, Chinese Nuthatch, Green-backed and Zappey’s Flycatchers and the rare Grey-sided Thrush. -

Adobe PDF, Job 6
Noms français des oiseaux du Monde par la Commission internationale des noms français des oiseaux (CINFO) composée de Pierre DEVILLERS, Henri OUELLET, Édouard BENITO-ESPINAL, Roseline BEUDELS, Roger CRUON, Normand DAVID, Christian ÉRARD, Michel GOSSELIN, Gilles SEUTIN Éd. MultiMondes Inc., Sainte-Foy, Québec & Éd. Chabaud, Bayonne, France, 1993, 1re éd. ISBN 2-87749035-1 & avec le concours de Stéphane POPINET pour les noms anglais, d'après Distribution and Taxonomy of Birds of the World par C. G. SIBLEY & B. L. MONROE Yale University Press, New Haven and London, 1990 ISBN 2-87749035-1 Source : http://perso.club-internet.fr/alfosse/cinfo.htm Nouvelle adresse : http://listoiseauxmonde.multimania. -

Bird Species I Have Seen World List
bird species I have seen U.K tally: 279 US tally: 393 Total world: 1,496 world list 1. Abyssinian ground hornbill 2. Abyssinian longclaw 3. Abyssinian white-eye 4. Acorn woodpecker 5. African black-headed oriole 6. African drongo 7. African fish-eagle 8. African harrier-hawk 9. African hawk-eagle 10. African mourning dove 11. African palm swift 12. African paradise flycatcher 13. African paradise monarch 14. African pied wagtail 15. African rook 16. African white-backed vulture 17. Agami heron 18. Alexandrine parakeet 19. Amazon kingfisher 20. American avocet 21. American bittern 22. American black duck 23. American cliff swallow 24. American coot 25. American crow 26. American dipper 27. American flamingo 28. American golden plover 29. American goldfinch 30. American kestrel 31. American mag 32. American oystercatcher 33. American pipit 34. American pygmy kingfisher 35. American redstart 36. American robin 37. American swallow-tailed kite 38. American tree sparrow 39. American white pelican 40. American wigeon 41. Ancient murrelet 42. Andean avocet 43. Andean condor 44. Andean flamingo 45. Andean gull 46. Andean negrito 47. Andean swift 48. Anhinga 49. Antillean crested hummingbird 50. Antillean euphonia 51. Antillean mango 52. Antillean nighthawk 53. Antillean palm-swift 54. Aplomado falcon 55. Arabian bustard 56. Arcadian flycatcher 57. Arctic redpoll 58. Arctic skua 59. Arctic tern 60. Armenian gull 61. Arrow-headed warbler 62. Ash-throated flycatcher 63. Ashy-headed goose 64. Ashy-headed laughing thrush (endemic) 65. Asian black bulbul 66. Asian openbill 67. Asian palm-swift 68. Asian paradise flycatcher 69. Asian woolly-necked stork 70.