WHO Drug Information Vol. 18, No. 4, 2004
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Présentation HJ
Conflits d’intérêts Astra-Zeneca, Janssen, Abacus international, Laboratoire ETAP, Institut Pasteur. Dr Hervé JAVELOT Pharmacien PH Etablissement Public de Santé Alsace Nord Service Pharmacie 141 avenue de Strasbourg 67 170 BRUMATH Tél. : 03 88 64 61 70 Fax : 03 88 64 61 58 Mail : [email protected] Perspectives dans la psychopharmacologie de l’anxiété et de la dépression Hervé JAVELOT Etablissement Public de Santé Alsace Nord Perspectives dans la psychopharmacologie de l’anxiété et de la dépression Traitements des troubles anxio-dépressifs. Les perspectives. ◦ L’axe GABAergique…éternellement prometteur ? ◦ L’incontournable théorie « monoaminergique »… ◦ Des théories alternatives à suivre … Traitements des troubles anxio-dépressifs. Contexte : ◦ XXI ème siècle : Le « siècle de la dépression » s’installe ? (Hardeveld et al., 2010) L’« ère de l’angoisse » s’affirme ? (Auden, 1947) ◦ Prévalence au cours de la vie : 16 à 17% pour la dépression, 17 à 18% pour les troubles anxieux Co-morbidité des 2 troubles dans 20 à 40% des cas (Antony, 2011 ; Depping et al., 2010 ; Hardeveld et al., 2010 ; Huppert, 2009) Auden WH (1947). The Age of Anxiety: A Baroque Eclogue. Random House: New York. Hardeveld F, Spijker J, De Graaf R, Nolen WA, Beekman AT. Prevalence and predictors of recurrence of major depressive disorder in the adult population. Acta Psychiatr Scand. 2010;122(3):184-91. Antony MM. Recent advances in the treatment of anxiety disorders. Canadian Psychology 2011;52(1), 10-19. Depping AM, Komossa K, Kissling W, Leucht S. Second-generation antipsychotics for anxiety disorders. Cochrane Database Syst Rev. 2010;(12):CD008120. Huppert JD. Anxiety disorders and depression comorbidity. -

Xenoport Hurt by Solzira NDA Withdrawal
November 11, 2008 Xenoport hurt by Solzira NDA withdrawal Evaluate Vantage News that Xenoport and partner GlaxoSmithKline’s recent application for restless legs syndrome (RLS) drug Solzira will have to be withdrawn, with the FDA requesting that data from a single trial be re-formatted, caused Xenoport’s shares to fall 13% yesterday to a new 18-month low of $34.44. With a potential 4-6 month delay to approval and the postponement of a $23m milestone that Xenoport was due to receive from Glaxo on the FDA’s acceptance of Solzira’s NDA, senior executives at the specialty US company could be forgiven for feeling somewhat aggrieved with Glaxo for this apparent oversight, given the pharma giant’s supposed experience and expertise in regulatory filings. The setback to Solzira and resulting slump in Xenoport’s share price suggests that shareholders who have not fled the stock will be even more desperate for positive phase IIb data for GERD treatment, XP19986 (Event - XenoPort looking for end of year trial lift, October 10, 2008). Formatting issues Avoiding these kinds of regulatory pitfalls, after all the cost and hard work involved in taking a drug through clinical development, is becoming an increasingly important factor behind a company’s decision to sign up a big pharma partner in today’s tougher regulatory environment. Both companies were naturally keen to stress that the NDA withdrawal has nothing to do with the content of the filing, which probably makes the rejection on a technicality all the more frustrating for Xenoport. Whilst the FDA requested the data from one particular study to be re-formatted, Glaxo has decided to review the formatting of other data sets in the application, presumably to ensure there can be no further grounds for rejection. -

PHARMACEUTICAL APPENDIX to the TARIFF SCHEDULE 2 Table 1
Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 36/84 (2006.01) A61K 31/5513 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/045 (2006.01) A61P 31/22 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/522 (2006.01) A61K 45/06 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/NL20 14/050780 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 13 November 2014 (13.1 1.2014) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/903,430 13 November 2013 (13. 11.2013) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: RJG DEVELOPMENTS B.V. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Translocator Protein (TSPO): the New Story of the Old Protein in Neuroinflammation
BMB Rep. 2020; 53(1): 20-27 BMB www.bmbreports.org Reports Invited Mini Review Translocator protein (TSPO): the new story of the old protein in neuroinflammation Younghwan Lee1, Youngjin Park1, Hyeri Nam1, Ji-Won Lee1 & Seong-Woon Yu1.2,* 1Department of Brain and Cognitive Sciences, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, 2Neurometabolomics Research Center, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Korea Translocator protein (TSPO), also known as peripheral Mitochondrial membrane proteins are the important regulators benzodiazepine receptor, is a transmembrane protein located of mitochondrial homeostasis, such as ion transport, ATP/ADP on the outer mitochondria membrane (OMM) and mainly transport, and mitochondria fusion/fission. Therefore, defects expressed in glial cells in the brain. Because of the close in these proteins are associated with numerous diseases (4). correlation of its expression level with neuropathology and For this reason, mitochondrial membrane proteins are therapeutic efficacies of several TSPO binding ligands under considered to be promising targets for development of diagnostic many neurological conditions, TSPO has been regarded as tools and therapeutic strategies. TSPO is the mitochondrial both biomarker and therapeutic target, and the biological transmembrane protein that was discovered as the binding site functions of TSPO have been a major research focus. of benzodiazepine in 1977 (5). Studies using TSPO-binding However, recent genetic studies with animal and cellular ligands have revealed that this protein participates in a variety models revealed unexpected results contrary to the anticipated of cellular functions, including cholesterol transport and biological importance of TSPO and cast doubt on the action steroid hormone synthesis, mitochondrial permeability transition modes of the TSPO-binding drugs. -

Serotonin Dopamine Antagonists)
NewNew DevelopmentsDevelopments inin thethe ResearchResearch andand TreatmentTreatment ofof SchizophreniaSchizophrenia StephenStephen M.M. Stahl,Stahl, MD,MD, PhDPhD Adjunct Professor, Department of Psychiatry, University of California, San Diego School of Medicine Sponsored by Neuroscience Education Institute Additionally sponsored by American Society for the Advancement of Pharmacotherapy This activity is supported by educational grants from AstraZeneca Pharmaceuticals LP; Cephalon, Inc.; and Shire Pharmaceuticals Inc. with additional support from Alkermes, Inc.; Eli Lilly and Company; Jazz Pharmaceuticals, Inc.; and Sepracor Inc. Copyright © 2007 Neuroscience Education Institute. All rights reserved. Learning Objectives After completion of this lecture you should be able to: • Evaluate evidence about the function and efficacy of the newest antipsychotics • Describe the rationale and present state of pharmacogenetics research • Discuss goals and challenges of pharmacogenetics in psychiatry Copyright © 2008 Neuroscience Education Institute. All rights reserved. Overview: Four Bedtime Stories • A Tale of Serotonin Antagonism • The Dopamine Partial Agonism Story • Pharmacogenetics and the Genes • Once Upon a Glutamate Receptor Copyright © 2008 Neuroscience Education Institute. All rights reserved. Overview: Four Bedtime Stories • A Tale of Serotonin Antagonism • The Dopamine Partial Agonism Story • Pharmacogenetics and the Genes • Once Upon a Glutamate Receptor Copyright © 2008 Neuroscience Education Institute. All rights reserved. What -

2009 Paris, France the Movement Disorder Society’S 13Th International Congress of Parkinson’S Disease and Movement Disorders
FINAL PROGRAM The Movement Disorder Society’s 13th International Congress OF PARKINSon’S DISEASE AND MOVEMENT DISORDERS JUNE 7-11, 2009 Paris, France The Movement Disorder Society’s 13th International Congress of Parkinson’s Disease and Movement Disorders Claiming CME Credit To claim CME credit for your participation in the MDS 13th International Congress of Parkinson’s Disease and Movement Disorders, International Congress participants must complete and submit an online CME Request Form. This Form will be available beginning June 10. Instructions for claiming credit: • After June 10, visit www.movementdisorders.org/congress/congress09/cme • Log in following the instructions on the page. You will need your International Congress Reference Number, located on the upper right of the Confirmation Sheet found in your registration packet. • Follow the on-screen instructions to claim CME Credit for the sessions you attended. • You may print your certificate from your home or office, or save it as a PDF for your records. Continuing Medical Education The Movement Disorder Society is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians. Credit Designation The Movement Disorder Society designates this educational activity for a maximum of 30.5 AMA PRA Category 1 Credits™. Physicians should only claim credit commensurate with the extent of their participation in the activity. Non-CME Certificates of Attendance were included with your on- site registration packet. If you did not receive one, please e-mail [email protected] to request one. The Movement Disorder Society has sought accreditation from the European Accreditation Council for Continuing Medical Education (EACCME) to provide the following CME activity for medical specialists. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

WO 2015/072853 Al 21 May 2015 (21.05.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/072853 Al 21 May 2015 (21.05.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 45/06 (2006.01) A61K 31/5513 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/045 (2006.01) A61K 31/5517 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/522 (2006.01) A61P 31/22 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 31/551 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/NL20 14/050781 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 13 November 2014 (13.1 1.2014) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 61/903,433 13 November 2013 (13. -
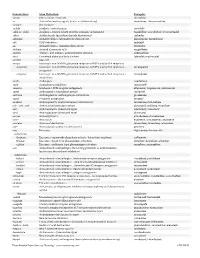
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

(12) Patent Application Publication (10) Pub. No.: US 2016/0220580 A1 Rubin Et Al
US 2016O220580A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0220580 A1 Rubin et al. (43) Pub. Date: Aug. 4, 2016 (54) SMALL MOLECULESCREENING FOR (60) Provisional application No. 61/497,708, filed on Jun. MOUSE SATELLITE CELL PROLIFERATION 16, 2011. (71) Applicant: PRESIDENT AND FELLOWS OF Publication Classification HARVARD COLLEGE, Cambridge, (51) Int. Cl. MA (US) A 6LX3/553 (2006.01) (72) Inventors: Lee L. Rubin, Wellesley, MA (US); A613 L/496 (2006.01) Amanda Gee, Alexandria, VA (US); A613 L/4439 (2006.01) Amy J. Wagers, Cambridge, MA (US) A613 L/404 (2006.01) (52) U.S. Cl. CPC ............. A6 IK3I/553 (2013.01); A61 K3I/404 (21) Appl. No.: 15/012,656 (2013.01); A61 K3I/496 (2013.01); A61 K 31/4439 (2013.01) (22) Filed: Feb. 1, 2016 (57) ABSTRACT The invention provides methods for inducing, enhancing or Related U.S. Application Data increasing satellite cell proliferation, and an assay for screen (63) Continuation-in-part of application No. 14/126,716, ing for a candidate compound for inducing, enhancing or filed on Jun. 13, 2014, now Pat. No. 9.248,185, filed as increasing satellite cell proliferation. Also provided are meth application No. PCT/US2012/042964 on Jun. 18, ods for repairing or regenerating a damaged muscle tissue of 2012. a Subject. Patent Application Publication Aug. 4, 2016 Sheet 1 of 44 US 2016/0220580 A1 FIG. A Patent Application Publication Aug. 4, 2016 Sheet 2 of 44 US 2016/0220580 A1 FIG. C. FIG. 2A Patent Application Publication Aug.