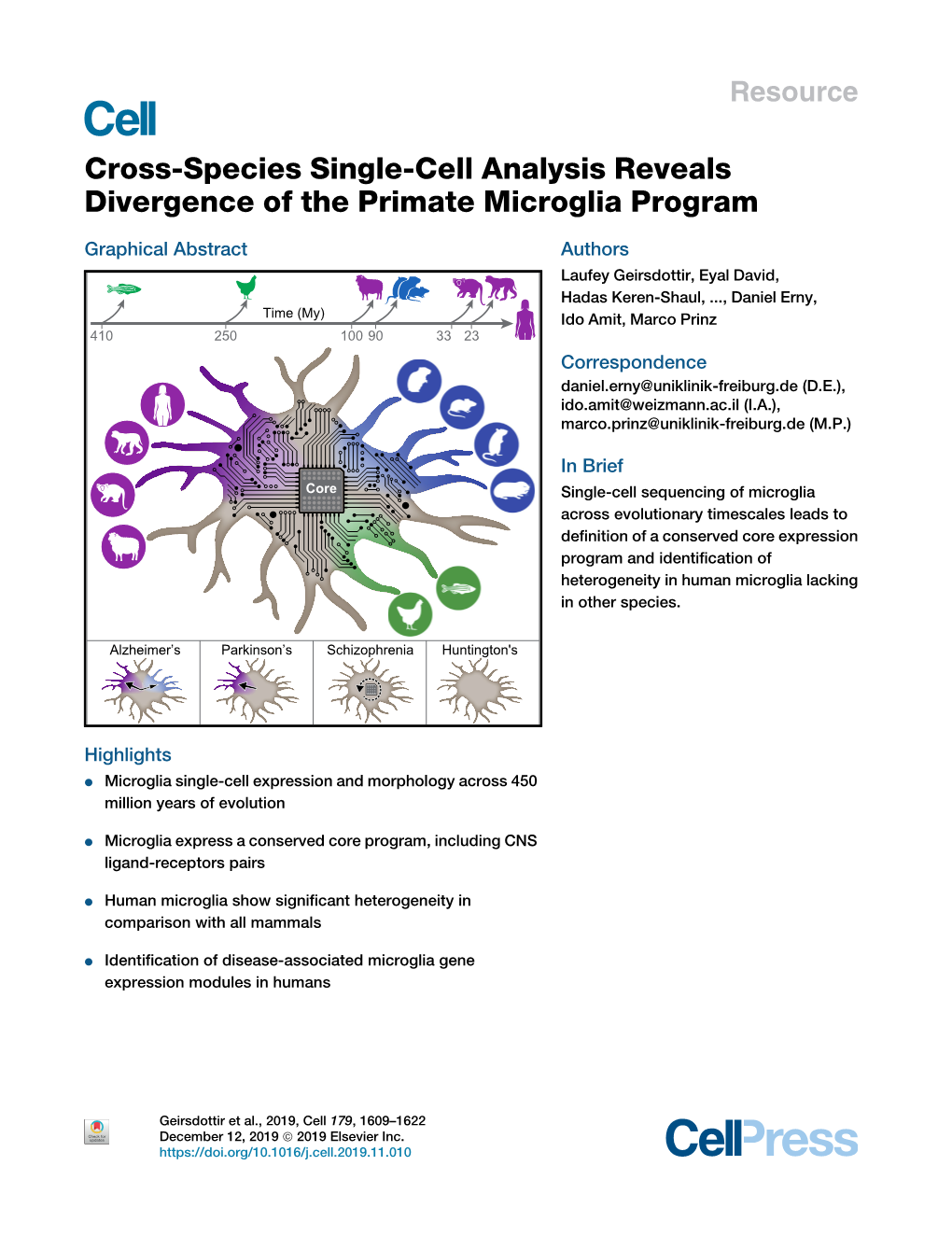
Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Signal Adaptor DAP10 Associates with MDL-1 and Triggers Osteoclastogenesis in Cooperation with DAP12
Signal adaptor DAP10 associates with MDL-1 and triggers osteoclastogenesis in cooperation with DAP12 Masanori Inuia,1, Yuki Kikuchia,1, Naoko Aokib, Shota Endoa, Tsutomu Maedaa, Akiko Sugahara-Tobinaia, Shion Fujimuraa, Akira Nakamuraa, Atsushi Kumanogohc, Marco Colonnad, and Toshiyuki Takaia,2 aDepartment of Experimental Immunology, Institute of Development, Aging and Cancer, Tohoku University, Seiryo 4-1, Sendai 980-8575, Japan; bDepartment of Pathology, Asahikawa Medical College, Higashi 2-1-1, Midorigaoka, Asahikawa 078-8510, Japan; cDepartment of Immunopathology, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka 565-0871, Japan; and dDepartment of Pathology and Immunology, Washington University School of Medicine, 660 South Euclid, St. Louis, MO 63110 Communicated by Jeffrey V. Ravetch, The Rockfeller University, New York, NY, January 22, 2009 (received for review May 14, 2008) Osteoclasts, cells of myeloid lineage, play a unique role in bone the Ig superfamily or C-type lectin family. They primarily mediate resorption, maintaining skeletal homeostasis in concert with bone- a Src-family kinase-initiated activation signal, leading to a series of producing osteoblasts. Osteoclast development and maturation (os- events including Syk kinase recruitment, triggering of calcium teoclastogenesis) is driven by receptor activator of NF-B ligand and signaling mediated by phospholipase C␥ (PLC␥), and activation of macrophage-colony stimulating factor and invariably requires a sig- the MAPK cascade, culminating in cell proliferation and cytokine nal initiated by immunoreceptor tyrosine-based activation motif production as the output of cellular responses. In osteoclast pre- (ITAM)-harboring Fc receptor common ␥ chain or DNAX-activating cursor cells, FcR␥ and DAP12 associate with several cognate protein (DAP)12 (also referred to as KARAP or TYROBP) that associates cell-surface immunoreceptors (9) such as osteoclast-associated with the cognate immunoreceptors. -

Distinct Gene Expression Pathways in Islets from Individuals with Short- and Long
Mastracci Teresa (Orcid ID: 0000-0003-1956-1951) Distinct Gene Expression Pathways in Islets from Individuals with Short- and Long- Duration Type 1 Diabetes Teresa L. Mastracci1,2*, Jean-Valery Turatsinze3, Benita K. Book4, Ivan A. Restrepo5, Michael J. Pugia1, Eric A. Wiebke4, Mark D. Pescovitz4, Decio L. Eizirik3 and Raghavendra G. Mirmira2,5,6,7 1 Indiana Biosciences Research Institute, Indianapolis, IN, USA. 2 Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, USA. 3 ULB Center for Diabetes Research, Universite Libre de Bruxelles, Brussels, Belgium. 4 Department of Surgery, Indiana University School of Medicine, Indianapolis, IN, USA. 5 Center for Diabetes and Metabolic Diseases, Indiana University School of Medicine, Indianapolis, IN, USA. 6 Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, USA. 7 Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA. * Corresponding Author: Teresa L. Mastracci, 1345 W. 16th St., Suite 300, Indianapolis, IN 46202 USA. E-mail: [email protected] Running Title: Functional Genomics Analysis of T1D Islets Abstract word count: 272 This is the author's manuscript of the article published in final edited form as: Mastracci, T. L., Turatsinze, J.-V., Book, B. K., Restrepo, I. A., Pugia, M. J., Wiebke, E. A., … Mirmira, R. G. (n.d.). Distinct Gene Expression Pathways in Islets from Individuals with Short‐ and Long‐Duration Type 1 Diabetes. Diabetes, Obesity and Metabolism. https://doi.org/10.1111/dom.13298 Main text word count: 2730 References: 27; Tables: 2; Figures: 3 Abstract Aims: Our current understanding of the pathogenesis of type 1 diabetes (T1D) arose in large part from studies using the non-obese diabetic (NOD) mouse model. -

Supplementary Materials
Supplementary Materials Supplemental Figure S1. Distinct difference in expression of 576 sensome genes comparing cortex versus microglia. (A) This heatmap shows all 576 sensome candidate genes ordered by DE and with the left column shows if the gene is present in the “Hickman et al. sensome” Supplemental Figure S2. Mouse sensome and human sensome genes categorized by group. (A) Bar graph showing the number of mouse and human sensome genes per group (Cell-Cell Interactions, Chemokine and related receptors, Cytokine receptors, ECM receptors, Endogenous ligands receptors, sensors and transporters, Fc receptors, Pattern recognition and related receptors, Potential sensors but no known ligands and Purinergic and related receptors). Supplementary Figure S3. Overlap of ligands recognized by microglia sensome (A) Overlap between the ligands of the receptors from respectively human and mouse core sensome was shown using Venn Diagrams. (B) Ligands of human and mouse receptors categorized in groups (Glycoproteins, Cytokines, Immunoglobulin, Amino acids, Carbohydrates, Electrolytes, Lipopeptides, Chemokines, Neuraminic acids, Nucleic acids, Receptors, Lipids, Fatty acids, Leukotrienes, Hormones, Steroids and Phospholipids) and spread of different groups shown as parts of whole again highlighting that the distribution of ligands what the human and mouse sensome genes can sense (Categorization of ligands in Supplementary Table S1). Supplementary Figure S4. Microglia core sensome expression during aging. (A) Two-log fold change of microglia core sensome genes in aging mice derived from Holtman et al. [12]. (B) Accelerated aging model (ERCC1), with impaired DNA repair mechanism, shows changes of microglia core sensome expression [12]. (C) Microglia core sensome expression during aging in human derived from Olah et al. -

A Transcriptomic Atlas of Aged Human Microglia
ARTICLE DOI: 10.1038/s41467-018-02926-5 OPEN A transcriptomic atlas of aged human microglia Marta Olah1,2, Ellis Patrick 3, Alexandra-Chloe Villani2,4, Jishu Xu 2, Charles C. White2, Katie J. Ryan5, Paul Piehowski 6, Alifiya Kapasi6, Parham Nejad2, Maria Cimpean 5, Sarah Connor1,2, Christina J. Yung1, Michael Frangieh5, Allison McHenry5, Wassim Elyaman1,2, Vlad Petyuk 6, Julie A. Schneider7, David A. Bennett7, Philip L. De Jager 1,2 & Elizabeth M. Bradshaw1,2 With a rapidly aging global human population, finding a cure for late onset neurodegenerative diseases has become an urgent enterprise. However, these efforts are hindered by the lack of 1234567890():,; understanding of what constitutes the phenotype of aged human microglia—the cell type that has been strongly implicated by genetic studies in the pathogenesis of age-related neuro- degenerative disease. Here, we establish the set of genes that is preferentially expressed by microglia in the aged human brain. This HuMi_Aged gene set captures a unique phenotype, which we confirm at the protein level. Furthermore, we find this gene set to be enriched in susceptibility genes for Alzheimer’s disease and multiple sclerosis, to be increased with advancing age, and to be reduced by the protective APOEε2 haplotype. APOEε4 has no effect. These findings confirm the existence of an aging-related microglial phenotype in the aged human brain and its involvement in the pathological processes associated with brain aging. 1 Center for Translational & Computational Neuroimmunology, Department of Neurology, Columbia University Medical Center, New York City, NY 10032, USA. 2 Program in Medical and Population Genetics, Broad Institute, Cambridge, MA 02142, USA. -

Transcriptomic Gene Signatures Associated with Persistent Airflow Limitation
Hekking et al Revised manuscript ERJ-02298-2016 Transcriptomic gene signatures associated with persistent airflow limitation in patients with severe asthma P.P. Hekking1, M.J. Loza2, S. Pavlidis3, B. De Meulder4, D. Lefaudeux4, F. Baribaud2, C. Auffray4, A.H. Wagener1, P. Brinkman1, R. Lutter1, A.T. Bansal5, A.R. Sousa6, S. Bates6, Y. Pandis3, L.J. Fleming3, D.E. Shaw7, S.J. Fowler8, Y. Guo3, A. Meiser3, K. Sun3, J. Corfield9, P. Howarth10, E.H. Bel1, I.M. Adcock11, K.F. Chung11, R. Djukanovic10, P.J. Sterk1, and the U-BIOPRED Study Group* 1: Respiratory Medicine, Academic Medical Centre, Amsterdam, the Netherlands; 2: Janssen research and development; Johnsen & Johnsen, Springhouse, US; 3: Data Science Institute, Imperial College, London, UK; 4: European Institute for Systems Biology and Medicine, CIRI UMR5308, CNRS-ENS- UCBL-INSERM, Université de Lyon, France; 5: Acclarogen Ltd, Cambridge, UK; 6: Discovery Medicine, GlaxoSmithKline, Brentford, UK; 7: Respiratory Research Unit, University of Nottingham, UK; 8: Centre for Respiratory Medicine and Allergy, Institute of Inflammation and Repair, University of Manchester and University Hospital of South Manchester, Manchester Academic Health Sciences Centre, UK; 9: Areteva, Nottingham, UK; 10: NIHR Southampton Centre for Biomedical Research, University of Southampton, UK; 11: National Heart & Lung institute - Imperial College, London, UK;*A list of the U-BIOPRED study group can be found in the online supplement Corresponding author: Pieter-Paul Hekking, Academic Medical Centre (AMC), Department of Respiratory Medicine, F5-260 1105 AZ, Amsterdam, the Netherlands Telephone: +31 (0) 20 566 1660 Fax: +31 (0) 20 566 9001 E-mail: [email protected] 1 Hekking et al Take home message Persistent airflow limitation in severe asthma is associated with a mechanism in which IL-13 and remodeling are involved. -

Supplementary Figure 1. Network Map Associated with Upregulated Canonical Pathways Shows Interferon Alpha As a Key Regulator
Supplementary Figure 1. Network map associated with upregulated canonical pathways shows interferon alpha as a key regulator. IPA core analysis determined interferon-alpha as an upstream regulator in the significantly upregulated genes from RNAseq data from nasopharyngeal swabs of COVID-19 patients (GSE152075). Network map was generated in IPA, overlaid with the Coronavirus Replication Pathway. Supplementary Figure 2. Network map associated with Cell Cycle, Cellular Assembly and Organization, DNA Replication, Recombination, and Repair shows relationships among significant canonical pathways. Significant pathways were identified from pathway analysis of RNAseq from PBMCs of COVID-19 patients. Coronavirus Pathogenesis Pathway was also overlaid on the network map. The orange and blue colors in indicate predicted activation or predicted inhibition, respectively. Supplementary Figure 3. Significant biological processes affected in brochoalveolar lung fluid of severe COVID-19 patients. Network map was generated by IPA core analysis of differentially expressed genes for severe vs mild COVID-19 patients in bronchoalveolar lung fluid (BALF) from scRNA-seq profile of GSE145926. Orange color represents predicted activation. Red boxes highlight important cytokines involved. Supplementary Figure 4. 10X Genomics Human Immunology Panel filtered differentially expressed genes in each immune subset (NK cells, T cells, B cells, and Macrophages) of severe versus mild COVID-19 patients. Three genes (HLA-DQA2, IFIT1, and MX1) were found significantly and consistently differentially expressed. Gene expression is shown per the disease severity (mild, severe, recovered) is shown on the top row and expression across immune cell subsets are shown on the bottom row. Supplementary Figure 5. Network map shows interactions between differentially expressed genes in severe versus mild COVID-19 patients. -

COVID-19: a Drug Repurposing and Biomarker Identification by Using Comprehensive Gene-Disease Associations Through Protein-Protein Interaction Network Analysis
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 30 March 2020 doi:10.20944/preprints202003.0440.v1 COVID-19: A drug repurposing and biomarker identification by using comprehensive gene-disease associations through protein-protein interaction network analysis Suresh Kumar* Department of Diagnostic and Allied Health Science, Faculty of Health & Life Sciences, Management & Science University, 40100 Shah Alam, Selangor, Malaysia *Corresponding author Suresh Kumar, PhD Department of Diagnostic and Allied Health Science, Faculty of Health & Life Sciences Management & Science University, 40100 Shah Alam, Selangor, Malaysia Email: [email protected] Abstract COVID-19 (2019-nCoV) is a pandemic disease with an estimated mortality rate of 3.4% (estimated by the WHO as of March 3, 2020). Until now there is no antiviral drug and vaccine for COVID-19. The current overwhelming situation by COVID-19 patients in hospitals is likely to increase in the next few months. About 15 percent of patients with serious disease in COVID-19 require immediate health services. Rather than waiting for new anti-viral drugs or vaccines that take a few months to years to develop and test, several researchers and public health agencies are attempting to repurpose medicines that are already approved for another similar disease and have proved to be fairly effective. This study aims to identify FDA approved drugs that can be used for drug repurposing and identify biomarkers among high- risk and asymptomatic groups. In this study gene-disease association related to COVID-19 reported mild, severe symptoms and clinical outcomes were determined. The high-risk group was studied related to SARS-CoV-2 viral entry and life cycle by using Disgenet and compared with curated COVID-19 gene data sets from the CTD database. -

During Intestinal Inflammation Functions Contributes to Regulatory
Reprogramming of Monocytes by GM-CSF Contributes to Regulatory Immune Functions during Intestinal Inflammation This information is current as Jan Däbritz, Toni Weinhage, Georg Varga, Timo Wirth, of October 2, 2021. Karoline Walscheid, Anne Brockhausen, David Schwarzmaier, Markus Brückner, Matthias Ross, Dominik Bettenworth, Johannes Roth, Jan M. Ehrchen and Dirk Foell J Immunol published online 4 February 2015 http://www.jimmunol.org/content/early/2015/02/04/jimmun ol.1401482 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/02/04/jimmunol.140148 Material 2.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 2, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published February 4, 2015, doi:10.4049/jimmunol.1401482 The Journal of Immunology Reprogramming of Monocytes by GM-CSF Contributes to Regulatory Immune Functions during Intestinal Inflammation Jan Da¨britz,*,†,‡,x,1 Toni Weinhage,*,1 Georg Varga,* Timo Wirth,* Karoline Walscheid,* Anne Brockhausen,{,‖ David Schwarzmaier,* Markus Bruckner,€ # Matthias Ross,# Dominik Bettenworth,# Johannes Roth,†,‖ Jan M. -

Signalling Through C-Type Lectin Receptors
REVIEWS Signalling through C-type lectin receptors: shaping immune responses Teunis B. H. Geijtenbeek and Sonja I. Gringhuis Abstract | C‑type lectin receptors (CLRs) expressed by dendritic cells are crucial for tailoring immune responses to pathogens. Following pathogen binding, CLRs trigger distinct signalling pathways that induce the expression of specific cytokines which determine T cell polarization fates. Some CLRs can induce signalling pathways that directly activate nuclear factor‑κB, whereas other CLRs affect signalling by Toll‑like receptors. Dissecting these signalling pathways and their effects on host immune cells is essential to understand the molecular mechanisms involved in the induction of adaptive immune responses. In this Review we describe the role of CLR signalling in regulating adaptive immunity and immunopathogenesis and discuss how this knowledge can be harnessed for the development of innovative vaccination approaches. Dendritic cells (DCs) are located throughout the body Triggering of several PRRs simultaneously can induce T helper (TH) cell A T cell subset that secretes a to capture and internalize invading pathogens, and diverse innate immune responses, which provides the distinct set of cytokines after subsequently process and present antigen on MHC diversity that is required to shape an effective adaptive activation, which occurs class I and class II molecules to CD8+ and CD4+ T cells, immune response. Distinct pathogens express different through the ligation of the T cell respectively1. Antigen presentation by DCs is in itself PAMPs, and the combination of these PAMPs functions receptor with its cognate ligand (peptide–MHC complex), not sufficient to induce effective T cell responses against as a fingerprint that triggers a specific set of PRRs, lead‑ + together with the recognition of pathogens. -

Transcriptional Profile of Human Anti-Inflamatory Macrophages Under Homeostatic, Activating and Pathological Conditions
UNIVERSIDAD COMPLUTENSE DE MADRID FACULTAD DE CIENCIAS QUÍMICAS Departamento de Bioquímica y Biología Molecular I TESIS DOCTORAL Transcriptional profile of human anti-inflamatory macrophages under homeostatic, activating and pathological conditions Perfil transcripcional de macrófagos antiinflamatorios humanos en condiciones de homeostasis, activación y patológicas MEMORIA PARA OPTAR AL GRADO DE DOCTOR PRESENTADA POR Víctor Delgado Cuevas Directores María Marta Escribese Alonso Ángel Luís Corbí López Madrid, 2017 © Víctor Delgado Cuevas, 2016 Universidad Complutense de Madrid Facultad de Ciencias Químicas Dpto. de Bioquímica y Biología Molecular I TRANSCRIPTIONAL PROFILE OF HUMAN ANTI-INFLAMMATORY MACROPHAGES UNDER HOMEOSTATIC, ACTIVATING AND PATHOLOGICAL CONDITIONS Perfil transcripcional de macrófagos antiinflamatorios humanos en condiciones de homeostasis, activación y patológicas. Víctor Delgado Cuevas Tesis Doctoral Madrid 2016 Universidad Complutense de Madrid Facultad de Ciencias Químicas Dpto. de Bioquímica y Biología Molecular I TRANSCRIPTIONAL PROFILE OF HUMAN ANTI-INFLAMMATORY MACROPHAGES UNDER HOMEOSTATIC, ACTIVATING AND PATHOLOGICAL CONDITIONS Perfil transcripcional de macrófagos antiinflamatorios humanos en condiciones de homeostasis, activación y patológicas. Este trabajo ha sido realizado por Víctor Delgado Cuevas para optar al grado de Doctor en el Centro de Investigaciones Biológicas de Madrid (CSIC), bajo la dirección de la Dra. María Marta Escribese Alonso y el Dr. Ángel Luís Corbí López Fdo. Dra. María Marta Escribese -

1 Supplemental Information Self-Sustaining Interleukin-8 Loops
Supplemental Information Self-sustaining interleukin-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19 Rainer Kaiser1,2,3*, Alexander Leunig1,2,*,§, Kami Pekayvaz1,2,3*, Oliver Popp4,5, Markus Joppich6, Vivien Polewka1, Raphael Escaig1, Afra Anjum1, Marie-Louise Hoffknecht1, Christoph Gold1, Sophia Brambs1, Anouk Engel1, Sven Stockhausen1,2, Viktoria Knottenberg1, Anna Titova1, Mohamed Haji4,5, Clemens Scherer1,2,3, Maximilian Muenchhoff3,8,9, Johannes C. Hellmuth3,10, Kathrin Saar4,5, Benjamin Schubert11,12,13, Anne Hilgendorff13,14,15, Christian Schulz1,2, Stefan Kääb1,2,3, Ralf Zimmer6, Norbert Hübner4,5,7, Steffen Massberg1,2,3, Philipp Mertins4,5, Leo Nicolai1,2,3,#,§, Konstantin Stark1,2,3,# Affiliations: 1 Department of Medicine I, University Hospital, Ludwig-Maximilians University Munich, Germany 2 DZHK (German Centre for Cardiovascular Research), partner site Munich Heart Alliance, Germany 3 COVID-19 Registry of the LMU Munich (CORKUM), University Hospital, Ludwig-Maximilians University Munich 4 Max Delbrück Center for Molecular Medicine (MDC) in the Helmholtz Association, Berlin, Germany 5 DZHK (German Centre for Cardiovascular Research), partner site Berlin, Germany 6 Department of Informatics, Ludwig-Maximilians-Universität München, Munich, Germany 7 Charité-Universitätsmedizin Berlin, Germany 8 Max von Pettenkofer Institute & GeneCenter, Virology, Faculty of Medicine, Ludwig-Maximilians University, Munich, Germany 9 German Center for Infection Research (DZIF), Partner Site Munich 10 Medizinische Klinik -

Genetic Loss-Of-Function of Activating Transcription Factor 3 but Not C-Type
www.nature.com/labinvest ARTICLE OPEN Genetic loss-of-function of activating transcription factor 3 but not C-type lectin member 5A prevents diabetic peripheral neuropathy ✉ Hung-Wei Kan1,6, Chin-Hong Chang2,6, Ying-Shuang Chang3, Yi-Ting Ko3 and Yu-Lin Hsieh 3,4,5 © The Author(s) 2021 We investigated the mediating roles of activating transcription factor 3 (ATF3), an injury marker, or C-type lectin member 5A (CLEC5A), an inflammatory response molecule, in the induction of endoplasmic reticulum (ER) stress and neuroinflammation in diabetic peripheral neuropathy in ATF3 and CLEC5A genetic knockout (aft3−/− and clec5a−/−, respectively) mice. ATF3 was expressed intranuclearly and was upregulated in mice with diabetic peripheral neuropathy (DN) and clec5a−/− mice. The DN and clec5a−/− groups also exhibited neuropathic behavior, but not in the aft3−/− group. The upregulation profiles of cytoplasmic polyadenylation element-binding protein, a protein translation–regulating molecule, and the ER stress-related molecules of inositol- requiring enzyme 1α and phosphorylated eukaryotic initiation factor 2α in the DN and clec5a−/− groups were correlated with neuropathic behavior. Ultrastructural evidence confirmed ER stress induction and neuroinflammation, including microglial enlargement and proinflammatory cytokine release, in the DN and clec5a−/− mice. By contrast, the induction of ER stress and neuroinflammation did not occur in the aft3−/− mice. Furthermore, the mRNA of reactive oxygen species–removing enzymes such as superoxide dismutase, heme oxygenase-1, and catalase were downregulated in the DN and clec5a−/− groups but were not changed in the aft3−/− group. Taken together, the results indicate that intraneuronal ATF3, but not CLEC5A, mediates the induction of ER stress and neuroinflammation associated with diabetic neuropathy.