Long-Term Remission of Severe Refractory Dermatopolymyositis With
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Percutaneous Conchotome Muscle Biopsy. a Useful Diagnostic and Assessment Tool CHRISTINA DORPH, INGER NENNESMO, and INGRID E
Percutaneous Conchotome Muscle Biopsy. A Useful Diagnostic and Assessment Tool CHRISTINA DORPH, INGER NENNESMO, and INGRID E. LUNDBERG ABSTRACT. Objective. To evaluate the diagnostic yield, performance simplicity, and safety of the percutaneous conchotome muscle biopsy technique for clinical and research purposes in an outpatient rheuma- tology clinic. Methods. Biopsies taken by rheumatologists in an outpatient clinic during 1996 and 1997 were eval- uated for histopathological and clinical diagnoses. Results. A total of 149 biopsies were performed on 122 patients. Physicians learned the method easily. Samples were of adequate size and quality to allow for diagnostics. In total 106 biopsies were taken due to different diagnostic suspicions: 24 polymyositis (PM) or dermatomyositis (DM); 43 PM, DM, or vasculitis in addition to another rheumatic condition; 19 systemic vasculitis; and 20 myalgias. Criteria for definite or probable PM/DM were fulfilled in 21 patients, 18 with positive biopsies. Thirteen patients received vasculitis as clinical diagnosis, 3 with positive biopsies. No patient with myalgia had a biopsy with inflammatory changes. Fifteen of 43 rebiopsies performed to assess disease activity had signs of active inflammation. In 48% there were changes in immuno- suppressive therapy after biopsy results. Four complications occurred; one was a serious subfascial hematoma. Conclusion. The percutaneous conchotome muscle biopsy technique gives a good size sample that allows for diagnostic evaluation and has a high yield in patients with myositis. It is a simple proce- dure, easy to learn and to perform, with a low complication rate and minimum discomfort for the patient. The method can preferably be used as a diagnostic tool and to perform repeated biopsies to assess the effect of a given therapy for both clinical and research purposes. -

Diagnosis and Treatment of Dermatomyositis-Systemic Lupus
Diagnosis and Treatment of Dermatomyositis-Systemic Lupus Erythematosus Overlap Syndrome Preston Williams1; Benjamin McKinney, MD2 1Texas A&M College of Medicine; 2Baylor University Medical Center Family Medicine Residency Introduction Case Description Discussion Dermatomyositis is an autoimmune condition classically A punch biopsy of her rash showed atrophic epithelium with This case of overlap syndrome between dermatomyositis and characterized by symmetric proximal muscle weakness, dyskeratotic keratinocytes, vacuolar interface changes, superficial systemic lupus erythematosus presents a rare but important inflammatory muscle changes, and dermatologic abnormalities.1 perivascular and lichenoid inflammation, and pigment challenge to the primary care physician. Our patient presented Several studies have shown that the inflammatory myopathies, incontinence consistent with systemic lupus erythematosus initially with arthralgias and fatigue, symptoms more such as dermatomyositis, commonly overlap with other (SLE). The patient was started on prednisone 40 mg daily for a 2- characteristic of systemic lupus erythematosus. However, these connective tissue disorders, significantly complicating the week taper to 10 mg and hydroxychloroquine 200 mg daily with symptoms were followed by a facial rash that involved the diagnosis.2 The reported incidence of overlap syndrome ranges marked improvement in symptoms. Further lab work-up was nasolabial folds and periorbital regions more in line with from 11% to 40% in patients diagnosed with significant -
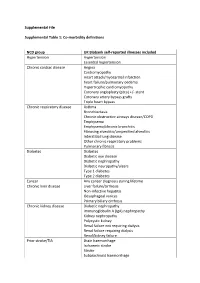
Supplemental File Supplemental Table 1: Co-Morbidity Definitions
Supplemental File Supplemental Table 1: Co-morbidity definitions NCD group UK Biobank self-reported illnesses included Hypertension Hypertension Essential hypertension Chronic cardiac disease Angina Cardiomyopathy Heart attack/myocardial infarction heart failure/pulmonary oedema Hypertrophic cardiomyopathy Coronary angioplasty (ptca) +/- stent Coronary artery bypass grafts Triple heart bypass Chronic respiratory disease Asthma Bronchiectasis Chronic obstructive airways disease/COPD Emphysema Emphysema/chronic bronchitis Fibrosing alveolitis/unspecified alveolitis Interstitial lung disease Other chronic respiratory problems Pulmonary fibrosis Diabetes Diabetes Diabetic eye disease Diabetic nephropathy Diabetic neuropathy/ulcers Type 1 diabetes Type 2 diabetes Cancer Any cancer diagnosis during lifetime Chronic liver disease Liver failure/cirrhosis Non-infective hepatitis Oesophageal varices Primary biliary cirrhosis Chronic kidney disease Diabetic nephropathy Immunoglobulin A (IgA) nephropathy Kidney nephropathy Polycystic kidney Renal failure not requiring dialysis Renal failure requiring dialysis Renal/kidney failure Prior stroke/TIA Brain haemorrhage Ischaemic stroke Stroke Subarachnoid haemorrhage Transient ischaemic attack (TIA) Other neurology disease Cerebral palsy Epilepsy Motor neurone disease Multiple sclerosis Myasthenia gravis Parkinson’s disease Psychiatric disease Depression Mania/bipolar disorder/manic depression Postnatal depression Schizophrenia Chronic inflammatory and Ankylosing spondylitis autoimmune rheumatic disease -

Idiopathic Inflammatory Myopathy
Idiopathic Infl ammatory Myopathy: Treatment Options Stephen J. DiMartino, MD, PhD Corresponding author ing PM or IBM can be more diffi cult [ 2 ]. The following Stephen J. DiMartino, MD, PhD noninfl ammatory myopathies and neurologic conditions Weill Medical College, Cornell University; and Hospital for Special Surgery, 535 East 70th Street, New York, NY 10021. can also present with proximal muscle weakness: cen- E-mail: [email protected] tral and peripheral nervous system disorders, adult-onset Current Rheumatology Reports 2008, 10: 321– 327 muscular dystrophies (eg, limb-girdle muscular dystro- Current Medicine Group LLC ISSN 1523-3774 phy, fascioscapulohumeral muscular dystrophy, Becker’s Copyright © 2008 by Current Medicine Group LLC muscular dystrophy), metabolic myopathies (eg, phospho- fructokinase defi ciency, acid-maltase defi ciency, carnitine palmityl-transferase II defi ciency, mitochondrial myopa- Idiopathic infl ammatory myopathy (IIM) comprises a thies), endocrine myopathies (eg, hypo- or hyperthyroidism, group of rare disorders in which there is an immune- Cushing’s syndrome, acromegaly), neuromuscular junction mediated attack on skeletal muscle, the consequence of disorders (eg, myasthenia gravis, Eaton-Lambert syndrome), which is muscle damage and weakness in the patient. viral myopathy, and toxic myopathy. As in other infl ammatory diseases, the general approach Today, statin myopathy is frequently considered in to therapy is use of immunosuppressive agents. the differential diagnosis of weakness or myalgia given Many options exist for IIM treatment, but therapeutic these agents’ high use in clinical practice [ 3 ]. Differen- approaches are based mostly on empirical evidence tiation among these entities often can be accomplished and small studies, many of which are uncontrolled. -

A Case of Dermatomyositis with Secondary Sjögren's Syndrome
162 A Case of Dermatomyositis with Secondary Sjögren’s Syndrome- Diagnosis with Follow-up Study of Technetium-99m Pyrophosphate Scintigraphy Ching-Tang Huang1, Ying-Chu Chen1, Chingtsai Lin2, Yu-Chun Hsiao3, Lai-Fa Sheu4, Min-Chien Tu1 Abstract Purpose: To report a case of dermatomyositis (DM) with secondary Sjögren’s syndrome (SS) and propose the clinical application of technetium-99m pyrophosphate (99mTc-PYP) scan. Case Report: A 50-year-old woman had progressive proximal muscle weakness of bilateral thighs, myalgia, tea-colored urine, and exercise intolerance for 6 months. Physical examination showed malar rash, V-sign, periungual erythema, and mechanic hands. Neurological assessment showed symmetric pelvic-girdle weakness, myopathic face, waddling gait, but preserved deep tendon reflex and sensory functions. DM was diagnosed on the basis of typical rashes and serum creatinine kinase elevation (7397 IU/L). Aside from myopathic symptoms, dry eye and mouth were reported. Thorough autoantibody searches showed positive anti-SSA/Ro antibody (198 U/ml). Both Schirmer's test and sialoscintigraphy were positive, leading secondary SS as diagnosis. Initial 99mTc-PYP scan revealed increased radiouptake in the muscles of bilateral thighs, compatible with clinical assessment. Follow- up scan three months later shows abnormal but attenuated radiouptake at bilateral thighs, in the presence of nearly-complete clinical recovery. Conclusion: DM with secondary SS in adult is a unique disease entity, with predominantly myopathic symptoms and satisfactory therapeutic response as its characteristics. Our serial muscle imaging studies suggest that 99mTc-PYP scan is at once anatomically-specific and persistently-sensitive to microstructural damages within inflammatory muscles, enabling clinician to monitor disease activity and therapeutic response. -

Document Downloaded from Day 24/07
Document downloaded from http://www.elsevier.es, day 27/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Document downloaded from http://www.elsevier.es, day 27/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Supplementary Table. Search strategy PUBMED #P1 "Idiopathic inflammatory myopathy" OR "autoimmune myopathy" OR myositis OR 205200 myopathy OR dermatomyositis OR "clinically amyopathic dermatomyositis" OR "amyopathic dermatomyositis" OR "hypomyopathic dermatomyositis" OR "juvenile dermatomyositis" OR "clinically amyopathic juvenile dermatomyositis" OR dermatopolymyositis OR polymyositis OR "immune-mediated necrotizing myopathy" OR "inclusion body myositis" OR "overlap myositis" OR "anti-synthetase syndrome" OR "cancer-associated myositis" #P2 Africa [MeSH] OR Africa [tw] OR Algeria [tw] OR Angola [tw] OR Benin [tw] OR 568425 Botswana [tw] OR Burkina Faso[tw] OR Burundi [tw] OR Cameroon [tw] OR Canary Islands [tw] OR Cape Verde [tw] OR Central African Republic [tw] OR Chad [tw] OR Comoros [tw] OR Congo [tw] OR Democratic Republic of Congo [tw] OR Djibouti [tw] OR Egypt [tw] OR Equatorial Guinea [tw] OR Eritrea [tw] OR Ethiopia [tw] OR Gabon [tw] OR Gambia [tw] OR Ghana [tw] OR Guinea [tw] OR Guinea Bissau [tw] OR Ivory Coast [tw] OR Cote d’Ivoire [tw] OR Kenya [tw] OR Lesotho [tw] OR Liberia [tw] OR Libya [tw] OR Libia [tw] OR Madagascar [tw] OR Malawi [tw] OR Mali [tw] OR Mauritania [tw] OR -

For Peer Review Hudaifa Alani¹, Julian R
Scandinavian Journal of Rheumatology Systematic review and meta-analysis of the epidemiology of polyautoimmunity in Sjögren’s syndrome (secondary Sjögren’s syndrome)For Peer focusing Review on autoimmune rheumatic diseases Journal: Scandinavian Journal of Rheumatology Manuscript ID SJR-2016-0514.R2 Manuscript Type: Article Date Submitted by the Author: 18-Apr-2017 Complete List of Authors: Alani, Hudaifa; Kettering General Hospital NHS Foundation Trust Henty, Julian; University College London Thompson, Nicolyn; University College London Jury, EC; University College London Ciurtin, Coziana; University College London, Department of Rheumatology secondary Sjogren's syndrome, epidemiology, Meta-analysis, systematic Keywords: review, polyautoimunity Scandinavian Journal of Rheumatology Page 1 of 71 Scandinavian Journal of Rheumatology Systematic review and meta-analysis of the epidemiology of polyautoimmunity in Sjögren’s syndrome (secondary Sjögren’s syndrome) focusing on autoimmune rheumatic diseases Submission type: article For Peer Review Short title: Polyautoimmunity in Sjögren's syndrome Authors: Hudaifa Alani¹, Julian R. Henty², Nicolyn L Thompson³, Elizabeth Jury³ and Coziana Ciurtin³* ¹Kettering General Hospital, NN16 8UZ, Kettering, United Kingdom ²Department of Medical Physics, University College London, London, United Kingdom ³Department of Rheumatology, University College London, London, United Kingdom Scandinavian Journal of Rheumatology Scandinavian Journal of Rheumatology Page 2 of 71 *Corresponding author: Dr. Coziana Ciurtin, PhD, FRCP, Department of Rheumatology, University College London, 250 Euston Road, London, NW1 2PG, email: [email protected], phone: +44(0)2034479035, fax: +44(0)20344797268 . For Peer Review Scandinavian Journal of Rheumatology Page 3 of 71 Scandinavian Journal of Rheumatology Abstract Objective: The epidemiology of polyautoimmunity in Sjögren's syndrome (secondary Sjögren's syndrome - sSS) is not well-defined and was not investigated before using a systematic approach. -

Chronic Intestinal Pseudo-Obstruction in Systemic Lupus Erythematosus Gut: First Published As 10.1136/Gut.43.1.117 on 1 July 1998
Gut 1998;43:117–122 117 Chronic intestinal pseudo-obstruction in systemic lupus erythematosus Gut: first published as 10.1136/gut.43.1.117 on 1 July 1998. Downloaded from G Perlemuter, S Chaussade, B Wechsler, P Cacoub, M Dapoigny, A Kahan, P Godeau, D Couturier Abstract enteric nerves, or the visceral autonomic nerv- Background/Aims—Chronic intestinal ous system. It may be the primary disease or it pseudo-obstruction (CIPO) reflects a dys- may be secondary to a recognised underlying function of the visceral smooth muscle or disease and then defined as secondary CIPO. the enteric nervous system. Gastrointesti- The causes of secondary CIPO are numerous nal manifestations are common in systemic and involve the nervous, endocrine, and meta- lupus erythematosus (SLE) but CIPO has bolic systems, intra-abdominal inflammation, not been reported. Features of CIPO are infiltrative and connective tissue diseases, and reported in five patients with SLE. drug induced states.2 Mild gastrointestinal Methods—From 1988 to 1993, five patients manifestations are common in systemic lupus with SLE or SLE-like syndrome were hos- erythematosus (SLE). Nausea, vomiting, diar- pitalised for gastrointestinal manometric rhoea, and abdominal pain are found in more studies. CIPO was the onset feature in two than 50% of these patients.3 Motility disorders cases. Antroduodenal manometry (three of the gastrointestinal tract are less frequent. hours fasting, two hours fed) was per- CIPO is rare in SLE. Since our first published formed in all patients, and oesophageal case,4 four other patients with SLE have been manometry in four. hospitalised in our department with a diagnosis Results—Intestinal hypomotility associ- of CIPO, confirmed by manometry. -

Dermatomyositis and Polymyositis
ARTIGO DE REVISÃO Dermatomiosite e polimiosite: da imunopatologia à imunoterapia (imunobiológicos) Samuel Katsuyuki Shinjo1, Fernando Henrique Carlos de Souza2, Julio Cesar Bertacini de Moraes2 RESUMO As miopatias infl amatórias idiopáticas (MII), das quais fazem parte a dermatomiosite (DM) e a polimiosite (PM), são doenças sistêmicas crônicas associadas a alta morbidade e incapacidade funcional. O tratamento atual baseia-se na corticoterapia e no uso de imunossupressores, porém uma parcela considerável dos pacientes é refratária à terapia tradicional. Isso tem levado à tentativa de uso de imunobiológicos nesses pacientes, tendo por fundamento a fi siopatogênese das MII. Do ponto de vista imunopatológico, há diferenças entre PM e DM: a primeira está mais relacionada à imunidade celular, enquanto na segunda o papel humoral parece mais importante. Em ambas, porém, são descritas concentrações elevadas de interleucinas pró-infl amatórias (TNF, IL-1, IL-6) e aumento da expressão de moléculas relacionadas à coestimulação dos linfócitos T – nessas condições, parece racional o uso da terapia biológica. Considerando os imunobiológicos disponíveis, são escassos os dados de trabalhos abertos na literatura, compostos principalmente por séries e relatos de casos. Os bloqueadores do TNF apresentam resultados confl itantes sem evidência de boa resposta ao tratamento. A terapia anti-CD20 possui os resultados mais promissores. É extremamente escassa a informação sobre o bloqueio da coestimulação do linfócito T e a terapia anti- IL-6, que impede qualquer consideração. Dessa maneira, o uso de imunobiológicos em MII ainda permanece como fronteira a ser explorada. A terapia biológica pode ter papel relevante no tratamento das MII refratárias à terapia convencional; no entanto, novos estudos prospectivos com base em parâmetros objetivos de resposta ao tratamento são necessários. -

B Disorders of Autonomic Nervous System
Application of the International Classification of Diseases to Neurology \ Second Edition World Health Organization Geneva 1997 First edition 1987 Second edition 1997 Application of the international classification of diseases to neurology: ICD-NA- 2nd ed. 1. Neurology - classification 2. Nervous system diseases - classification I. Title: ICD-NA ISBN 92 4 154502 X (NLM Classification: WL 15) The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available. ©World Health Organization 1997 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Convention. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturers' products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of -

Systemic Lupus Erythematosus- Myositis Overlap
Clinics and Practice 2011; volume 1:e89 Systemic lupus erythematosus- same patient. 1 Myositis (polymyositis PM or dermatomyositis DM) identifies a group of Correspondence: Faten Frikha, Department of myositis overlap syndrome: patients in whom the mascular weakness is Internal Medicine, Hospital of Hedi Chaker, 3029 report of 6 cases the principle clinical feature often associated Sfax, Tunisia. with muscle pain, tenderness and wasting, or E-mail: [email protected] Fatma Maazoun, 1 Faten Frikha, 1 other form of connective tissue diseases; the Key words: overlap syndrome, systemic lupus ery - Mouna Snoussi, 1 Neila Kaddour, 1 muscle biopsy generally demonstrates areas of thematosus, myositis, classification criteria, cor - Hatem Masmoudi, 2 Zouhir Bahloul 1 muscle fibre necrosis accompanied by intersti - ticosteroid. tial and/or perivascular cellular infiltrates. 1Department of Internal Medicine, Received for publication: 22 September 2011. Myositis associated with overlap syndromes is Hospital of Hedi Chaker; 2Laboratory of Accepted for publication: 12 October 2011. usually of paroxysmal variety and has been immunology, Hospital Habib Bourguiba, associated with one or another of connective This work is licensed under a Creative Commons Sfax, Tunisia tissue disorders [Systemic Sclerosis (SSc), Attribution NonCommercial 3.0 License (CC BY- Rheumatoid arthritis (RA), Sjögren’s syn - NC 3.0). drome or systemic lupus erythematous (SLE)]. ©Copyright F. Maazoun et al., 2011 Pearson and Bohan found an incidence of 21% Licensee PAGEPress, Italy Abstract 2 of this type of myositis. Myositis is a rare com - Clinics and Practice 2011; 1:e89 plication of systemic lupus erythematous 3,4 doi:10.4081/cp.2011.e89 The incidence of myositis in patients with occurring in almost 4-16% of cases of SLE 3,5,6 systemic lupus erythematosus (SLE) is low and such association is considered to be an among different series. -

Guidelines of the Brazilian Society of Rheumatology for the Treatment Of
Souza et al. Advances in Rheumatology (2019) 59:6 Advances in Rheumatology https://doi.org/10.1186/s42358-019-0048-x POSITION ARTICLE AND GUIDELINES Open Access Guidelines of the Brazilian Society of Rheumatology for the treatment of systemic autoimmune myopathies Fernando Henrique Carlos de Souza1, Daniel Brito de Araújo2, Verônica Silva Vilela3, Mailze Campos Bezerra4, Ricardo Santos Simões1, Wanderley Marques Bernardo1, Renata Miossi1, Bernardo Matos da Cunha5 and Samuel Katsuyuki Shinjo6* Abstract Background: Recommendations of the Myopathy Committee of the Brazilian Society of Rheumatology for the management and therapy of systemic autoimmune myopathies (SAM). Main body: The review of the literature was done in the search for the Medline (PubMed), Embase and Cochrane databases including studies published until June 2018. The Prisma was used for the systematic review and the articles were evaluated according to the levels of Oxford evidence. Ten recommendations were developed addressing the management and therapy of systemic autoimmune myopathies. Conclusions: Robust data to guide the therapeutic process are scarce. Although not proven effective in controlled clinical trials, glucocorticoid represents first-line drugs in the treatment of SAM. Intravenous immunoglobulin is considered in induction for refractory cases of SAM or when immunosuppressive drugs are contra-indicated. Consideration should be given to the early introduction of immunosuppressive drugs. There is no specific period determined for the suspension of glucocorticoid and immunosuppressive drugs when individually evaluating patients with SAM. A key component for treatment in an early rehabilitation program is the inclusion of strength- building and aerobic exercises, in addition to a rigorous evaluation of these activities for remission of disease and the education of the patient and his/her caregivers.