Tau Spreading Mechanisms; Implications for Dysfunctional Tauopathies
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Autosomal Dominant Leukodystrophy with Autonomic Symptoms
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I MR imaging characteristics and neuropathology of the spin- al cord in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. Sundblom J, Melberg A, Kalimo H, Smits A, Raininko R. AJNR Am J Neuroradiol. 2009 Feb;30(2):328-35. II Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Schuster J, Sundblom J, Thuresson AC, Hassin-Baer S, Klopstock T, Dichgans M, Cohen OS, Raininko R, Melberg A, Dahl N. Neurogenetics. 2011 Feb;12(1):65-72. III Bedside diagnosis of rippling muscle disease in CAV3 p.A46T mutation carriers. Sundblom J, Stålberg E, Osterdahl M, Rücker F, Montelius M, Kalimo H, Nennesmo I, Islander G, Smits A, Dahl N, Melberg A. Muscle Nerve. 2010 Jun;41(6):751-7. IV A family with discordance between Malignant hyperthermia susceptibility and Rippling muscle disease. Sundblom J, Mel- berg A, Rücker F, Smits A, Islander G. Manuscript submitted to Journal of Anesthesia. Reprints were made with permission from the respective publishers. Contents Introduction..................................................................................................11 Background and history............................................................................... 13 Early studies of hereditary disease...........................................................13 Darwin and Mendel................................................................................ -

Follow-Up Study of 22 Chinese Children with Alexander Disease and Analysis of Parental Origin of De Novo GFAP Mutations
Journal of Human Genetics (2013) 58, 183–188 & 2013 The Japan Society of Human Genetics All rights reserved 1434-5161/13 www.nature.com/jhg ORIGINAL ARTICLE Follow-up study of 22 Chinese children with Alexander disease and analysis of parental origin of de novo GFAP mutations Lili Zang1, Jingmin Wang1, Yuwu Jiang1, Qiang Gu1, Zhijie Gao1, Yanling Yang1, Jiangxi Xiao2 and Ye Wu1 To delineate the phenotype and genotype in Chinese children with type I Alexander disease (AxD) and the parental origin of de novo glial fibrillary acidic protein (GFAP) mutations. Twenty-two children with clinically diagnosed type I AxD were followed up for 1.66–6.62 years. Allele-specific PCR was used for the analysis of parental origin of the allele harboring the de novo mutation. Phenotype of these patients were consistent with type I AxD described in other population, with developmental delay (motor delay in 81.82%, cognitive delay in 63.64%), macrocephaly (100%), seizures (95.45%), paroxysmal deterioration (27.27%) and typical brain magnetic resonance imaging (100%). Progression was slower than reported. At 8.55 years of age (5.29–13.25), all patients who underwent the second follow-up were alive. Eleven heterozygous missense mutations of GFAP were identified in 21 patients, with three novel mutations. Reported hot spot mutations, p.R79, p.R239 and p.R88, were also identified in Chinese patients. Mutations were de novo in all but one case. The mother of a proband was demonstrated to be a presymptomatic patient with type II AxD with a p.R79H mutation. Ninety percent of de novo mutations were on the paternal allele demonstrated by allele-specific PCR. -

Infantile Alexander Disease: Case Report and Review of Literature Dentistry Section
Case Report DOI: 10.7860/JCDR/2017/26875.10106 Infantile Alexander Disease: Case Report and Review of Literature Dentistry Section SOUMYABRATA SARKAR1, RUPAM SINHA2, AMITABHA CHAKRABORTY3, TANYA KHAITAN4, BIYAS BHOWMIK5 ABSTRACT Alexander Disease (AD) is an autosomal dominant leukodystrophy and occurs predominantly in infants and children. It usually results in death within ten years after onset. Among the four subtypes, infantile form comprises the most of affected individuals. It presents in the first two years of life, typically with progressive psychomotor deficiency, loss of developmental milestones, seizures, and pyramidal signs. Clinical and magnetic resonance image findings usually establish diagnosis of AD. Here, we present a case of Infantile AD with characteristic clinical and radiological features. Keywords: Leukodystrophy, Magnetic resonance imaging, Seizures CASE REPORT A five-year-old boy reported to the Department of Oral Medicine and Radiology, with painful decayed tooth in the upper right back jaw region for 10 days. The pain was mild, continuous, non- radiating in nature with no associated factors. The patient’s parents gave history of delayed developmental milestones of their son. There was no history of consanguineous marriage of the patient’s parents. They reported their son to be normal at birth with standard birth weight and head circumference. There was history of seizures six months after birth, with gradual psychomotor deficiencies and cognitive abnormalities, for which his parents sought paediatric consultation immediately at seven months of age. Initial consultation was inconclusive due to lack of diagnostic evidence and financial constrictions. However, he was put on phenytoin oral suspension twice daily, which controlled the symptoms. -

Alexander Disease HH 2020 LCN Winter Meeting
2020 Winter Meeting March 3, 2020 Alexander Disease Speakers • CHOP : Katy McDonald, MSN,CRNP • Family Advocate : De Flohre Alexander Disease Children What is Alexander Disease? • Referred to as ALX, AxD, Demyelinogenic Leukodystrophy, Fibrinoid Leukodystrophy, White Matter Disease, and other names • Alexander Disease is a fatal neurodegenerative disorder that destroys structure and functionality of neurons in the brain • Demyelination caused by lack of Glial Fibrillary Acidic Protein (GFAP) • Only leukodystrophy in which Rosenthal Fibers appear • Rosenthal Fibers are abnormal protein deposits that cause improper cell function Rosenthal fibers Too much GFAP Gain of Mice (couresy of Albee Messing) function protein Morphologically and biochemically identical to human Rosenthal fibers How do you get Alexander Disease? In our cohort at CHOP, • Typically Genetic approximately 90% • 95% of patients have a mutation in GFAP. No other genes have Type I (younger) patients have a de been associated with AxD. novo mutation on • Mosaic parents Chromosome 17 that • Later-onset is classically genetic expresses Glial • autosomal dominant form Fibrillary Acidic Protein (GFAP) GFAP gene How Does Alexander Disease Affect the Individual? • Alexander Disease is a demyelinating disease and causes the loss of many functions such as walking, talking, and eating • Alexander Disease is progressive and causes severe loss of previously attained milestones or failure to thrive in the infantile form What are Initial Symptoms of Alexander Disease? The Different Types of Alexander’ Disease Present Differently Patients with juvenile • alexander’s disease are Macrocephaly sometimes misdiagnosed with • Seizures anorexia due to the severity of • Developmental Delays their failure to thrive and • Vomiting, swallowing or feeding issues feeding issues. -

Protein Aggregate Myopathies
Review Article Protein aggregate myopathies M. C. Sharma, H. H. Goebel1 All India Institute of Medical Sciences, New Delhi, India and 1Department of Neuropathology, Johannes Gutenberg University Medical Center, Mainz, Germany Protein aggregate myopathies (PAM) are an emerging group Aggegation of proteins within muscle cells may be of lyso- of muscle diseases characterized by structural abnormali- somal or nonlysosomal nature. The former occurs, for instance, ties. Protein aggregate myopathies are marked by the ag- in the neuronal ceroid-lipofuscinoses (NCL) as components gregation of intrinsic proteins within muscle fibers and fall of lipopigments, their accretion being a morphological hall- into four major groups or conditions: (1) desmin-related mark in the NCL.[1] This lysosomal accumulation of myopathies (DRM) that include desminopathies, a-B lipopigments renders the NCL a member of the lysosomal dis- crystallinopathies, selenoproteinopathies caused by muta- orders among which type-II glycogenosis, Niemann-Pick dis- tions in the, a-B crystallin and selenoprotein N1 genes, (2) ease, mucolipidosis IV and others may affect the skeletal hereditary inclusion body myopathies, several of which have muscle. been linked to different chromosomal gene loci, but with as Nonlysosomal accumulation of proteins within muscle fibers yet unidentified protein product, (3) actinopathies marked is a hallmark of a recently emerging subgroup of myopathies, by mutations in the sarcomeric ACTA1 gene, and (4) called protein aggregate myopathies (PAM). The cause -

Developing New Molecular Tools to Study and Visualize the Intermediate Filament GFAP in Living Cells
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS DEPARTAMENTO DE BIOLOGIA VEGETAL Developing new molecular tools to study and visualize the intermediate filament GFAP in living cells Ricardo Jorge Pinheiro Quiteres Mestrado em Biologia Molecular e Genética Dissertação orientada por: Professor Doutor Federico Herrera Doutor Zach Hensel 2019 Acknowledgments The completion of this thesis would not have been possible without the help, guidance and support of so, so many people: I start by thanking my supervisor Dr. Federico Herrera for welcoming me in your group, for your guid- ance through each stage of this thesis and for all the patience and time you spent teaching me. I would like to express my deepest gratitude to Dr. Zach Hensel for your tremendous help and advice, even when I was not technically your student. I am also thankful to Dr. Alvaro Crevenna for all the advice and for the knowledge that he shared. I also wish to thank the Flow Cytometry and the Advanced Imaging Facilities of Instituto Gulbenkian de Ciência for their services and assistance. Thank you to all the people from Fede’s lab, especially to three very talented PhD students. First to Cristina for all your help with that diabolic technique. To Fernanda thank you for all your help, encour- agement and support and for taking time off your experiments to teach me. Finally, to my mentor and friend Ricardo, thank you for your massive help, for teaching me basically everything that I know and for answering an enormous amount of questions and not answering a lot more, allowing me to think and grow as a scientist. -

Leukodystrophies by Raphael Schiffmann MD (Dr
Leukodystrophies By Raphael Schiffmann MD (Dr. Schiffmann, Director of the Institute of Metabolic Disease at Baylor Research Institute, received research grants from Amicus Therapeutics, Protalix Biotherapeutics, and Shire.) Originally released January 17, 2013; last updated November 25, 2016; expires November 25, 2019 Introduction Overview Leukodystrophies are a heterogeneous group of genetic disorders affecting the white matter of the central nervous system and sometimes with peripheral nervous system involvement. There are over 30 different leukodystrophies, with an overall population incidence of 1 in 7663 live births. They are now most commonly grouped based on the initial pattern of central nervous system white matter abnormalities on neuroimaging. All leukodystrophies have MRI hyperintense white matter on T2-weighted images, whereas T1 signal may be variable. Mildly hypo-, iso-, or hyperintense T1 signal relative to the cortex suggests a hypomyelinating pattern. A significantly hypointense T1 signal is more often associated with demyelination or other pathologies. Recognition of the abnormal MRI pattern in leukodystrophies greatly facilitates its diagnosis. Early diagnosis is important for genetic counseling and appropriate therapy where available. Key points • Leukodystrophies are classically defined as progressive genetic disorders that predominantly affect the white matter of the brain. • The pattern of abnormalities on brain MRI, and sometimes brain CT, is the most useful diagnostic tool. • Radial diffusivity on brain diffusion weighted imaging correlates with motor handicap. • X-linked adrenoleukodystrophy is the most common leukodystrophy and has effective therapy if applied early in the disease course. • Lentiviral hemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy shows promise. Historical note and terminology The first leukodystrophies were identified early last century. -
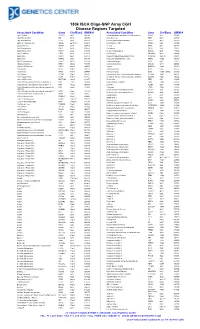
180K ISCA Oligo-SNP Array CGH Disease Regions Targeted
180k ISCA Oligo-SNP Array CGH Disease Regions Targeted Associated Condition Gene Chr/Band OMIM # Associated Condition Gene Chr/Band OMIM # 1p31 Deletion DIRAS3 1p31 605193 Chondrodysplasia punctata, X-linked recessive CDPX1 Xp22 302950 1p36 Microdeletion SKI 1p36 607872 Choroideremia CHM Xp21 303100 1p36 Microdeletion TP73 1p36 607872 Chronic granulomatous disease CYBB Xp11 306400 2p15-16.1 Microdeletion Multiple 2p15-16.1 612513 Chronic pancreatitis SPINK1 5q32 167800 2q37.3 Deletion HDAC4 2q37 600430 Cleft lip MSX1 4p16 608874 3q29 Microdeletion PAK2 3q29 609425 Cleft palate SATB2 2q32 119540 6p24 Microdeletion FKHL7 6p24 612852 Cleidocranial dysplasia RUNX2 6p21 119600 8p23.1 Deletion CTSB 8p23 116810 Coffin-Lowry RPS6KA3 Xp22 303600 9p Deletion DMRT1 9p24 158170 Congenital adrenal hyperplasia (CAH) CYP21A2 6p21.32 201910 9p Deletion DMRT2 9p24 158170 Congenital diaphragmatic hernia NR2F2 15q26 142340 9q34.3 Microdeletion EHMT1 9q34.3 610253 Cornelia de Lange 1 NIPBL 5p13 122470 10q22-23 Deletion GRID1 10q22 610659 Cornelia de Lange 2 SMC1L1 Xp11 300590 12q14.1-q15 Deletion GRIP1 12q14 604597 Cowden BMPR1A 10q23 158350 13q Deletion GPC5 13q31 602446 Craniosynostosis MSX2 5q35 604757 13q Deletion GPC6 13q31 604404 Craniosynostosis SOX6 11p15.1-p15.2 218350 13q Deletion PCDH9 13q21 603581 Creatine deficiency / X-linked mental retardation SLC6A8 16p11 300352 14q11-q22 Deletion CHD8 14q11 613457 Creatine deficiency / X-linked mental retardation SLC6A8 16p11 300352 14q11-q22 Deletion SUPT16H 14q11 613457 Cri-du-Chat TERT 5p15 123450 14q22 -

Charcot-Marie-Tooth and Giant Axonal
F1000Research 2014, 3:83 Last updated: 16 MAY 2019 OPINION ARTICLE Recommendations to enable drug development for inherited neuropathies: Charcot-Marie-Tooth and Giant Axonal Neuropathy [version 2; peer review: 2 approved] Lori Sames1, Allison Moore2,3, Renée J.G. Arnold4,5, Sean Ekins 1-4,6-9 1Hannah's Hope Fund, Rexford, NY, 12148, USA 2BioGAN Therapeutics, Rexford, NY, 12148, USA 3Hereditary Neuropathy Foundation, New York, NY, 10016, USA 4Arnold Consultancy & Technology LLC, New York, NY, 10023, USA 5Master of Public Health Program, Mount Sinai School of Medicine, New York, NY, 10029, USA 6Collaborations in Chemistry, Fuquay Varina, NC27526, USA 7Department of Pharmaceutical Sciences, University of Maryland, Baltimore, MD, 21201, USA 8Department of Pharmacology, Rutgers-Robert Wood Johnson Medical School, Piscataway, NJ, 08854, USA 9Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599-7355, USA First published: 03 Apr 2014, 3:83 ( Open Peer Review v2 https://doi.org/10.12688/f1000research.3751.1) Latest published: 22 Apr 2014, 3:83 ( https://doi.org/10.12688/f1000research.3751.2) Reviewer Status Abstract Invited Reviewers Approximately 1 in 2500 Americans suffer from Charcot-Marie-Tooth (CMT) 1 2 disease. The underlying disease mechanisms are unique in most forms of CMT, with many point mutations on various genes causing a toxic accumulation of misfolded proteins. Symptoms of the disease often present version 2 report within the first two decades of life, with CMT1A patients having reduced published compound muscle and sensory action potentials, slow nerve conduction 22 Apr 2014 velocities, sensory loss, progressive distal weakness, foot and hand deformities, decreased reflexes, bilateral foot drop and about 5% become version 1 wheelchair bound. -

Autophagy in Myelinating Glia
256 • The Journal of Neuroscience, January 8, 2020 • 40(2):256–266 Viewpoints Autophagy in Myelinating Glia Jillian Belgrad,1 XRaffaella De Pace,2 and R. Douglas Fields1 1Section on Nervous System Development and Plasticity and 2Section on Intracellular Protein Trafficking, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892 Autophagy is the cellular process involved in transportation and degradation of membrane, proteins, pathogens, and organelles. This fundamental cellular process is vital in development, plasticity, and response to disease and injury. Compared with neurons, little informationisavailableonautophagyinglia,butitisparamountforgliatoperformtheircriticalresponsestonervoussystemdiseaseand injury, including active tissue remodeling and phagocytosis. In myelinating glia, autophagy has expanded roles, particularly in phago- cytosis of mature myelin and in generating the vast amounts of membrane proteins and lipids that must be transported to form new myelin. Notably, autophagy plays important roles in removing excess cytoplasm to promote myelin compaction and development of oligodendrocytes, as well as in remyelination by Schwann cells after nerve trauma. This review summarizes the cell biology of autophagy, detailing the major pathways and proteins involved, as well as the roles of autophagy in Schwann cells and oligodendrocytes in develop- ment, plasticity, and diseases in which myelin is affected. This includes traumatic brain injury, Alexander’s -

Deep Phenotyping for Translational Research and Precision Medicine NIH Symposium: Linking Disease Model Phenotypes to Human Conditions
Deep Phenotyping for Translational Research and Precision Medicine NIH Symposium: Linking Disease Model Phenotypes to Human Conditions Peter Robinson Charit´e Universit¨atsmedizin Berlin September 10–11, 2015 Peter Robinson (Charite)´ Deep Phenotyping 1/50 September 10–11, 2015 1 / 50 Thanks! Matthew Brush Nathan Dunn Melissa Haendel Harry Hochheiser Sebastian K¨ohler Suzanna Lewis Julie McMurry Christopher Mungall Peter Robinson Damian Smedley Nicole Vasilevsky Kent Shefchek Nicole Washington Zhou Yuan 1 http://monarchinitiative.org Peter Robinson (Charite)´ Deep Phenotyping 2/50 September 10–11, 2015 2 / 50 Plan 1 Human Phenotype Ontology (HPO) 2 Ontology Algorithms: The Bare-Bones Basics 3 The Phenomizer 4 The HPO for translational research 5 PhenIX: Clinical Diagnostics in Medical Genetics 6 HPO: Semantic Unification of Common and Rare Disease 7 Pressing Needs and Goals for Future Impact Peter Robinson (Charite)´ Deep Phenotyping 3/50 September 10–11, 2015 3 / 50 Bioinformatics Since the beginnings of the field of Bioinformatics in the 1960s, a central theme has been the development of algorithms that calculate similarity scores between biological entities and use them to rank lists Margaret Dayhoff, originator of PAM matrices BLAST: Find and rank homologous sequences Peter Robinson (Charite)´ Deep Phenotyping 4/50 September 10–11, 2015 4 / 50 Bioinformatics for medicine? But how exactly do we calculate the similarity between diseases, symptoms, patients,:::? Peter Robinson (Charite)´ Deep Phenotyping 5/50 September 10–11, 2015 5 / 50 The Human Phenotype Ontology 11,030 terms 117,348 annotations for ∼ 7000 mainly monogenic diseases http://www.human-phenotype-ontology.org Widely used in rare disease community: UK 100,000 genomes; NIH Undiagnosed Diseases Network; DDD/DECIPHER, GA4GH, etc.