Multivariate Statistical Analysis Using the R Package Chemometrics
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Real-Time Wavelet Compression and Self-Modeling Curve
REAL-TIME WAVELET COMPRESSION AND SELF-MODELING CURVE RESOLUTION FOR ION MOBILITY SPECTROMETRY A dissertation presented to the faculty of the College of Arts and Sciences of Ohio University In partial fulfillment of the requirements for the degree Doctor of Philosophy Guoxiang Chen March 2003 This dissertation entitled REAL-TIME WAVELET COMPRESSION AND SELF-MODELING CURVE RESOLUTION FOR ION MOBILITY SPECTROMETRY BY GUOXIANG CHEN has been approved for the Department of Chemistry and Biochemistry and the College of Arts and Sciences by Peter de B. Harrington Associate Professor of Chemistry and Biochemistry Leslie A. Flemming Dean, College of Arts and Sciences CHEN, GUOXIANG. Ph.D. March 2003. Analytical Chemistry Real-Time Wavelet Compression and Self-Modeling Curve Resolution for Ion Mobility Spectrometry (203 pp.) Director of Dissertation: Peter de B. Harrington Chemometrics has proven useful for solving chemistry problems. Most of the chemometric methods are applied in post-run analyses, for which data are processed after being collected and archived. However, in many applications, real-time processing is required to obtain knowledge underlying complex chemical systems instantly. Moreover, real-time chemometrics can eliminate the storage burden for large amounts of raw data that occurs in post-run analyses. These attributes are important for the construction of portable intelligent instruments. Ion mobility spectrometry (IMS) furnishes inexpensive, sensitive, fast, and portable sensors that afford a wide variety of potential applications. SIMPLe-to-use Interactive Self-modeling Mixture Analysis (SIMPLISMA) is a self-modeling curve resolution method that has been demonstrated as an effective tool for enhancing IMS measurements. However, all of the previously reported studies have applied SIMPLISMA as a post-run tool. -

Curriculum Vitae
Jeff Goldsmith 722 W 168th Street, 6th floor New York, NY 10032 jeff[email protected] Date of Preparation April 20, 2021 Academic Appointments / Work Experience 06/2018{Present Department of Biostatistics Mailman School of Public Health, Columbia University Associate Professor 06/2012{05/2018 Department of Biostatistics Mailman School of Public Health, Columbia University Assistant Professor 01/2009{12/2010 Department of Biostatistics Bloomberg School of Public Health, Johns Hopkins University Research Assistant (R01NS060910) 01/2008{12/2009 Department of Biostatistics Bloomberg School of Public Health, Johns Hopkins University Research Assistant (U19 AI060614 and U19 AI082637) Education 08/2007{05/2012 Johns Hopkins University PhD in Biostatistics, May 2012 Thesis: Statistical Methods for Cross-sectional and Longitudinal Functional Observations Advisors: Ciprian Crainiceanu and Brian Caffo 08/2003{05/2007 Dickinson College BS in Mathematics, May 2007 Jeff Goldsmith 2 Honors 04/2021 Dean's Excellence in Leadership Award 03/2021 COPSS Leadership Academy For Emerging Leaders in Statistics 06/2017 Tow Faculty Scholar 01/2016 Public Voices Fellow 10/2013 Calderone Junior Faculty Prize 05/2012 ASA Biometrics Section Travel Award 12/2011 Invited Paper in \Highlights of JCGS" Session at Interface 05/2011 Margaret Merrell Award for Outstanding Research by a Biostatistics Doc- toral Student 05/2011 School-wide Teaching Assistant Recognition Award 05/2011 Helen Abbey Award for Excellence in Teaching 03/2011 ENAR Distinguished Student Paper Award 05/2010 Jane and Steve Dykacz Award for Outstanding Paper in Medical Statistics 05/2009 Nominated for School-wide Teaching Assistant Recognition Award 08/2007{05/2012 Sommer Scholar 05/2007 James Fowler Rusling Prize 05/2007 Lance E. -

Comparison of Harmonic, Geometric and Arithmetic Means for Change Detection in SAR Time Series Guillaume Quin, Béatrice Pinel-Puysségur, Jean-Marie Nicolas
Comparison of Harmonic, Geometric and Arithmetic means for change detection in SAR time series Guillaume Quin, Béatrice Pinel-Puysségur, Jean-Marie Nicolas To cite this version: Guillaume Quin, Béatrice Pinel-Puysségur, Jean-Marie Nicolas. Comparison of Harmonic, Geometric and Arithmetic means for change detection in SAR time series. EUSAR. 9th European Conference on Synthetic Aperture Radar, 2012., Apr 2012, Germany. hal-00737524 HAL Id: hal-00737524 https://hal.archives-ouvertes.fr/hal-00737524 Submitted on 2 Oct 2012 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. EUSAR 2012 Comparison of Harmonic, Geometric and Arithmetic Means for Change Detection in SAR Time Series Guillaume Quin CEA, DAM, DIF, F-91297 Arpajon, France Béatrice Pinel-Puysségur CEA, DAM, DIF, F-91297 Arpajon, France Jean-Marie Nicolas Telecom ParisTech, CNRS LTCI, 75634 Paris Cedex 13, France Abstract The amplitude distribution in a SAR image can present a heavy tail. Indeed, very high–valued outliers can be observed. In this paper, we propose the usage of the Harmonic, Geometric and Arithmetic temporal means for amplitude statistical studies along time. In general, the arithmetic mean is used to compute the mean amplitude of time series. -

Fast Computation of the Deviance Information Criterion for Latent Variable Models
Crawford School of Public Policy CAMA Centre for Applied Macroeconomic Analysis Fast Computation of the Deviance Information Criterion for Latent Variable Models CAMA Working Paper 9/2014 January 2014 Joshua C.C. Chan Research School of Economics, ANU and Centre for Applied Macroeconomic Analysis Angelia L. Grant Centre for Applied Macroeconomic Analysis Abstract The deviance information criterion (DIC) has been widely used for Bayesian model comparison. However, recent studies have cautioned against the use of the DIC for comparing latent variable models. In particular, the DIC calculated using the conditional likelihood (obtained by conditioning on the latent variables) is found to be inappropriate, whereas the DIC computed using the integrated likelihood (obtained by integrating out the latent variables) seems to perform well. In view of this, we propose fast algorithms for computing the DIC based on the integrated likelihood for a variety of high- dimensional latent variable models. Through three empirical applications we show that the DICs based on the integrated likelihoods have much smaller numerical standard errors compared to the DICs based on the conditional likelihoods. THE AUSTRALIAN NATIONAL UNIVERSITY Keywords Bayesian model comparison, state space, factor model, vector autoregression, semiparametric JEL Classification C11, C15, C32, C52 Address for correspondence: (E) [email protected] The Centre for Applied Macroeconomic Analysis in the Crawford School of Public Policy has been established to build strong links between professional macroeconomists. It provides a forum for quality macroeconomic research and discussion of policy issues between academia, government and the private sector. The Crawford School of Public Policy is the Australian National University’s public policy school, serving and influencing Australia, Asia and the Pacific through advanced policy research, graduate and executive education, and policy impact. -

University of Cincinnati
UNIVERSITY OF CINCINNATI Date:___________________ I, _________________________________________________________, hereby submit this work as part of the requirements for the degree of: in: It is entitled: This work and its defense approved by: Chair: _______________________________ _______________________________ _______________________________ _______________________________ _______________________________ Gibbs Sampling and Expectation Maximization Methods for Estimation of Censored Values from Correlated Multivariate Distributions A dissertation submitted to the Division of Research and Advanced Studies of the University of Cincinnati in partial ful…llment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (Ph.D.) in the Department of Mathematical Sciences of the McMicken College of Arts and Sciences May 2008 by Tina D. Hunter B.S. Industrial and Systems Engineering The Ohio State University, Columbus, Ohio, 1984 M.S. Aerospace Engineering University of Cincinnati, Cincinnati, Ohio, 1989 M.S. Statistics University of Cincinnati, Cincinnati, Ohio, 2003 Committee Chair: Dr. Siva Sivaganesan Abstract Statisticians are often called upon to analyze censored data. Environmental and toxicological data is often left-censored due to reporting practices for mea- surements that are below a statistically de…ned detection limit. Although there is an abundance of literature on univariate methods for analyzing this type of data, a great need still exists for multivariate methods that take into account possible correlation amongst variables. Two methods are developed here for that purpose. One is a Markov Chain Monte Carlo method that uses a Gibbs sampler to es- timate censored data values as well as distributional and regression parameters. The second is an expectation maximization (EM) algorithm that solves for the distributional parameters that maximize the complete likelihood function in the presence of censored data. -

A Generalized Linear Model for Binomial Response Data
A Generalized Linear Model for Binomial Response Data Copyright c 2017 Dan Nettleton (Iowa State University) Statistics 510 1 / 46 Now suppose that instead of a Bernoulli response, we have a binomial response for each unit in an experiment or an observational study. As an example, consider the trout data set discussed on page 669 of The Statistical Sleuth, 3rd edition, by Ramsey and Schafer. Five doses of toxic substance were assigned to a total of 20 fish tanks using a completely randomized design with four tanks per dose. Copyright c 2017 Dan Nettleton (Iowa State University) Statistics 510 2 / 46 For each tank, the total number of fish and the number of fish that developed liver tumors were recorded. d=read.delim("http://dnett.github.io/S510/Trout.txt") d dose tumor total 1 0.010 9 87 2 0.010 5 86 3 0.010 2 89 4 0.010 9 85 5 0.025 30 86 6 0.025 41 86 7 0.025 27 86 8 0.025 34 88 9 0.050 54 89 10 0.050 53 86 11 0.050 64 90 12 0.050 55 88 13 0.100 71 88 14 0.100 73 89 15 0.100 65 88 16 0.100 72 90 17 0.250 66 86 18 0.250 75 82 19 0.250 72 81 20 0.250 73 89 Copyright c 2017 Dan Nettleton (Iowa State University) Statistics 510 3 / 46 One way to analyze this dataset would be to convert the binomial counts and totals into Bernoulli responses. -

Comparison of Some Chemometric Tools for Metabonomics Biomarker Identification ⁎ Réjane Rousseau A, , Bernadette Govaerts A, Michel Verleysen A,B, Bruno Boulanger C
Available online at www.sciencedirect.com Chemometrics and Intelligent Laboratory Systems 91 (2008) 54–66 www.elsevier.com/locate/chemolab Comparison of some chemometric tools for metabonomics biomarker identification ⁎ Réjane Rousseau a, , Bernadette Govaerts a, Michel Verleysen a,b, Bruno Boulanger c a Université Catholique de Louvain, Institut de Statistique, Voie du Roman Pays 20, B-1348 Louvain-la-Neuve, Belgium b Université Catholique de Louvain, Machine Learning Group - DICE, Belgium c Eli Lilly, European Early Phase Statistics, Belgium Received 29 December 2006; received in revised form 15 June 2007; accepted 22 June 2007 Available online 29 June 2007 Abstract NMR-based metabonomics discovery approaches require statistical methods to extract, from large and complex spectral databases, biomarkers or biologically significant variables that best represent defined biological conditions. This paper explores the respective effectiveness of six multivariate methods: multiple hypotheses testing, supervised extensions of principal (PCA) and independent components analysis (ICA), discriminant partial least squares, linear logistic regression and classification trees. Each method has been adapted in order to provide a biomarker score for each zone of the spectrum. These scores aim at giving to the biologist indications on which metabolites of the analyzed biofluid are potentially affected by a stressor factor of interest (e.g. toxicity of a drug, presence of a given disease or therapeutic effect of a drug). The applications of the six methods to samples of 60 and 200 spectra issued from a semi-artificial database allowed to evaluate their respective properties. In particular, their sensitivities and false discovery rates (FDR) are illustrated through receiver operating characteristics curves (ROC) and the resulting identifications are used to show their specificities and relative advantages.The paper recommends to discard two methods for biomarker identification: the PCA showing a general low efficiency and the CART which is very sensitive to noise. -
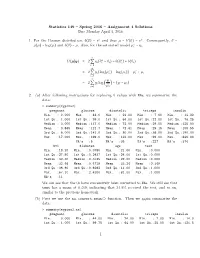
Statistics 149 – Spring 2016 – Assignment 4 Solutions Due Monday April 4, 2016 1. for the Poisson Distribution, B(Θ) =
Statistics 149 { Spring 2016 { Assignment 4 Solutions Due Monday April 4, 2016 1. For the Poisson distribution, b(θ) = eθ and thus µ = b′(θ) = eθ. Consequently, θ = ∗ g(µ) = log(µ) and b(θ) = µ. Also, for the saturated model µi = yi. n ∗ ∗ D(µSy) = 2 Q yi(θi − θi) − b(θi ) + b(θi) i=1 n ∗ ∗ = 2 Q yi(log(µi ) − log(µi)) − µi + µi i=1 n yi = 2 Q yi log − (yi − µi) i=1 µi 2. (a) After following instructions for replacing 0 values with NAs, we summarize the data: > summary(mypima2) pregnant glucose diastolic triceps insulin Min. : 0.000 Min. : 44.0 Min. : 24.00 Min. : 7.00 Min. : 14.00 1st Qu.: 1.000 1st Qu.: 99.0 1st Qu.: 64.00 1st Qu.:22.00 1st Qu.: 76.25 Median : 3.000 Median :117.0 Median : 72.00 Median :29.00 Median :125.00 Mean : 3.845 Mean :121.7 Mean : 72.41 Mean :29.15 Mean :155.55 3rd Qu.: 6.000 3rd Qu.:141.0 3rd Qu.: 80.00 3rd Qu.:36.00 3rd Qu.:190.00 Max. :17.000 Max. :199.0 Max. :122.00 Max. :99.00 Max. :846.00 NA's :5 NA's :35 NA's :227 NA's :374 bmi diabetes age test Min. :18.20 Min. :0.0780 Min. :21.00 Min. :0.000 1st Qu.:27.50 1st Qu.:0.2437 1st Qu.:24.00 1st Qu.:0.000 Median :32.30 Median :0.3725 Median :29.00 Median :0.000 Mean :32.46 Mean :0.4719 Mean :33.24 Mean :0.349 3rd Qu.:36.60 3rd Qu.:0.6262 3rd Qu.:41.00 3rd Qu.:1.000 Max. -

Bayesian Methods: Review of Generalized Linear Models
Bayesian Methods: Review of Generalized Linear Models RYAN BAKKER University of Georgia ICPSR Day 2 Bayesian Methods: GLM [1] Likelihood and Maximum Likelihood Principles Likelihood theory is an important part of Bayesian inference: it is how the data enter the model. • The basis is Fisher’s principle: what value of the unknown parameter is “most likely” to have • generated the observed data. Example: flip a coin 10 times, get 5 heads. MLE for p is 0.5. • This is easily the most common and well-understood general estimation process. • Bayesian Methods: GLM [2] Starting details: • – Y is a n k design or observation matrix, θ is a k 1 unknown coefficient vector to be esti- × × mated, we want p(θ Y) (joint sampling distribution or posterior) from p(Y θ) (joint probabil- | | ity function). – Define the likelihood function: n L(θ Y) = p(Y θ) | i| i=1 Y which is no longer on the probability metric. – Our goal is the maximum likelihood value of θ: θˆ : L(θˆ Y) L(θ Y) θ Θ | ≥ | ∀ ∈ where Θ is the class of admissable values for θ. Bayesian Methods: GLM [3] Likelihood and Maximum Likelihood Principles (cont.) Its actually easier to work with the natural log of the likelihood function: • `(θ Y) = log L(θ Y) | | We also find it useful to work with the score function, the first derivative of the log likelihood func- • tion with respect to the parameters of interest: ∂ `˙(θ Y) = `(θ Y) | ∂θ | Setting `˙(θ Y) equal to zero and solving gives the MLE: θˆ, the “most likely” value of θ from the • | parameter space Θ treating the observed data as given. -

What I Should Have Described in Class Today
Statistics 849 Clarification 2010-11-03 (p. 1) What I should have described in class today Sorry for botching the description of the deviance calculation in a generalized linear model and how it relates to the function dev.resids in a glm family object in R. Once again Chen Zuo pointed out the part that I had missed. We need to use positive contributions to the deviance for each observation if we are later to take the square roots of these quantities. To ensure that these are all positive we establish a baseline level for the deviance, which is the lowest possible value of the deviance, corresponding to a saturated model in which each observation has one parameter associated with it. Another value quoted in the summary output is the null deviance, which is the deviance for the simplest possible model (the \null model") corresponding to a formula like > data(Contraception, package = "mlmRev") > summary(cm0 <- glm(use ~ 1, binomial, Contraception)) Call: glm(formula = use ~ 1, family = binomial, data = Contraception) Deviance Residuals: Min 1Q Median 3Q Max -0.9983 -0.9983 -0.9983 1.3677 1.3677 Coefficients: Estimate Std. Error z value Pr(>|z|) (Intercept) -0.43702 0.04657 -9.385 <2e-16 (Dispersion parameter for binomial family taken to be 1) Null deviance: 2590.9 on 1933 degrees of freedom Residual deviance: 2590.9 on 1933 degrees of freedom AIC: 2592.9 Number of Fisher Scoring iterations: 4 Notice that the coefficient estimate, -0.437022, is the logit transformation of the proportion of positive responses > str(y <- as.integer(Contraception[["use"]]) - 1L) int [1:1934] 0 0 0 0 0 0 0 0 0 0 .. -

Multivariate Chemometrics As a Strategy to Predict the Allergenic Nature of Food Proteins
S S symmetry Article Multivariate Chemometrics as a Strategy to Predict the Allergenic Nature of Food Proteins Miroslava Nedyalkova 1 and Vasil Simeonov 2,* 1 Department of Inorganic Chemistry, Faculty of Chemistry and Pharmacy, University of Sofia, 1 James Bourchier Blvd., 1164 Sofia, Bulgaria; [email protected]fia.bg 2 Department of Analytical Chemistry, Faculty of Chemistry and Pharmacy, University of Sofia, 1 James Bourchier Blvd., 1164 Sofia, Bulgaria * Correspondence: [email protected]fia.bg Received: 3 September 2020; Accepted: 21 September 2020; Published: 29 September 2020 Abstract: The purpose of the present study is to develop a simple method for the classification of food proteins with respect to their allerginicity. The methods applied to solve the problem are well-known multivariate statistical approaches (hierarchical and non-hierarchical cluster analysis, two-way clustering, principal components and factor analysis) being a substantial part of modern exploratory data analysis (chemometrics). The methods were applied to a data set consisting of 18 food proteins (allergenic and non-allergenic). The results obtained convincingly showed that a successful separation of the two types of food proteins could be easily achieved with the selection of simple and accessible physicochemical and structural descriptors. The results from the present study could be of significant importance for distinguishing allergenic from non-allergenic food proteins without engaging complicated software methods and resources. The present study corresponds entirely to the concept of the journal and of the Special issue for searching of advanced chemometric strategies in solving structural problems of biomolecules. Keywords: food proteins; allergenicity; multivariate statistics; structural and physicochemical descriptors; classification 1. -

Copula-Based Analysis of Dependent Data with Censoring and Zero Inflation
Copula-Based Analysis of Dependent Data with Censoring and Zero Inflation by Fuyuan Li B.S. in Telecommunication Engineering, May 2012, Beijing University of Technology M.S. in Statistics, May 2014, The George Washington University A Dissertation submitted to The Faculty of The Columbian College of Arts and Sciences of The George Washington University in partial satisfaction of the requirements for the degree of Doctor of Philosophy January 10, 2019 Dissertation directed by Huixia J. Wang Professor of Statistics The Columbian College of Arts and Sciences of The George Washington University certifies that Fuyuan Li has passed the Final Examination for the degree of Doctor of Philosophy as of December 7, 2018. This is the final and approved form of the dissertation. Copula-Based Analysis of Dependent Data with Censoring and Zero Inflation Fuyuan Li Dissertation Research Committee: Huixia J. Wang, Professor of Statistics, Dissertation Director Tapan K. Nayak, Professor of Statistics, Committee Member Reza Modarres, Professor of Statistics, Committee Member ii c Copyright 2019 by Fuyuan Li All rights reserved iii Acknowledgments This work would not have been possible without the financial support of the National Science Foundation grant DMS-1525692, and the King Abdullah University of Science and Technology office of Sponsored Research award OSR-2015-CRG4-2582. I am grateful to all of those with whom I have had the pleasure to work during this and other related projects. Each of the members of my Dissertation Committee has provided me extensive personal and professional guidance and taught me a great deal about both scientific research and life in general.