Varmap: a Web Tool for Mapping Genomic Coordinates to Protein Sequence and Structure and Retrieving Protein Structural Annotations James D
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Uniprot at EMBL-EBI's Role in CTTV
Barbara P. Palka, Daniel Gonzalez, Edd Turner, Xavier Watkins, Maria J. Martin, Claire O’Donovan European Bioinformatics Institute (EMBL-EBI), European Molecular Biology Laboratory, Wellcome Genome Campus, Hinxton, Cambridge, CB10 1SD, UK UniProt at EMBL-EBI’s role in CTTV: contributing to improved disease knowledge Introduction The mission of UniProt is to provide the scientific community with a The Centre for Therapeutic Target Validation (CTTV) comprehensive, high quality and freely accessible resource of launched in Dec 2015 a new web platform for life- protein sequence and functional information. science researchers that helps them identify The UniProt Knowledgebase (UniProtKB) is the central hub for the collection of therapeutic targets for new and repurposed medicines. functional information on proteins, with accurate, consistent and rich CTTV is a public-private initiative to generate evidence on the annotation. As much annotation information as possible is added to each validity of therapeutic targets based on genome-scale experiments UniProtKB record and this includes widely accepted biological ontologies, and analysis. CTTV is working to create an R&D framework that classifications and cross-references, and clear indications of the quality of applies to a wide range of human diseases, and is committed to annotation in the form of evidence attribution of experimental and sharing its data openly with the scientific community. CTTV brings computational data. together expertise from four complementary institutions: GSK, Biogen, EMBL-EBI and Wellcome Trust Sanger Institute. UniProt’s disease expert curation Q5VWK5 (IL23R_HUMAN) This section provides information on the disease(s) associated with genetic variations in a given protein. The information is extracted from the scientific literature and diseases that are also described in the OMIM database are represented with a controlled vocabulary. -

Webnetcoffee
Hu et al. BMC Bioinformatics (2018) 19:422 https://doi.org/10.1186/s12859-018-2443-4 SOFTWARE Open Access WebNetCoffee: a web-based application to identify functionally conserved proteins from Multiple PPI networks Jialu Hu1,2, Yiqun Gao1, Junhao He1, Yan Zheng1 and Xuequn Shang1* Abstract Background: The discovery of functionally conserved proteins is a tough and important task in system biology. Global network alignment provides a systematic framework to search for these proteins from multiple protein-protein interaction (PPI) networks. Although there exist many web servers for network alignment, no one allows to perform global multiple network alignment tasks on users’ test datasets. Results: Here, we developed a web server WebNetcoffee based on the algorithm of NetCoffee to search for a global network alignment from multiple networks. To build a series of online test datasets, we manually collected 218,339 proteins, 4,009,541 interactions and many other associated protein annotations from several public databases. All these datasets and alignment results are available for download, which can support users to perform algorithm comparison and downstream analyses. Conclusion: WebNetCoffee provides a versatile, interactive and user-friendly interface for easily running alignment tasks on both online datasets and users’ test datasets, managing submitted jobs and visualizing the alignment results through a web browser. Additionally, our web server also facilitates graphical visualization of induced subnetworks for a given protein and its neighborhood. To the best of our knowledge, it is the first web server that facilitates the performing of global alignment for multiple PPI networks. Availability: http://www.nwpu-bioinformatics.com/WebNetCoffee Keywords: Multiple network alignment, Webserver, PPI networks, Protein databases, Gene ontology Background tools [7–10] have been developed to understand molec- Proteins are involved in almost all life processes. -

The Biogrid Interaction Database
D470–D478 Nucleic Acids Research, 2015, Vol. 43, Database issue Published online 26 November 2014 doi: 10.1093/nar/gku1204 The BioGRID interaction database: 2015 update Andrew Chatr-aryamontri1, Bobby-Joe Breitkreutz2, Rose Oughtred3, Lorrie Boucher2, Sven Heinicke3, Daici Chen1, Chris Stark2, Ashton Breitkreutz2, Nadine Kolas2, Lara O’Donnell2, Teresa Reguly2, Julie Nixon4, Lindsay Ramage4, Andrew Winter4, Adnane Sellam5, Christie Chang3, Jodi Hirschman3, Chandra Theesfeld3, Jennifer Rust3, Michael S. Livstone3, Kara Dolinski3 and Mike Tyers1,2,4,* 1Institute for Research in Immunology and Cancer, Universite´ de Montreal,´ Montreal,´ Quebec H3C 3J7, Canada, 2The Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada, 3Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, NJ 08544, USA, 4School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3JR, UK and 5Centre Hospitalier de l’UniversiteLaval´ (CHUL), Quebec,´ Quebec´ G1V 4G2, Canada Received September 26, 2014; Revised November 4, 2014; Accepted November 5, 2014 ABSTRACT semi-automated text-mining approaches, and to en- hance curation quality control. The Biological General Repository for Interaction Datasets (BioGRID: http://thebiogrid.org) is an open access database that houses genetic and protein in- INTRODUCTION teractions curated from the primary biomedical lit- Massive increases in high-throughput DNA sequencing erature for all major model organism species and technologies (1) have enabled an unprecedented level of humans. As of September 2014, the BioGRID con- genome annotation for many hundreds of species (2–6), tains 749 912 interactions as drawn from 43 149 pub- which has led to tremendous progress in the understand- lications that represent 30 model organisms. -

The Uniprot Knowledgebase BLAST
Introduction to bioinformatics The UniProt Knowledgebase BLAST UniProtKB Basic Local Alignment Search Tool A CRITICAL GUIDE 1 Version: 1 August 2018 A Critical Guide to BLAST BLAST Overview This Critical Guide provides an overview of the BLAST similarity search tool, Briefly examining the underlying algorithm and its rise to popularity. Several WeB-based and stand-alone implementations are reviewed, and key features of typical search results are discussed. Teaching Goals & Learning Outcomes This Guide introduces concepts and theories emBodied in the sequence database search tool, BLAST, and examines features of search outputs important for understanding and interpreting BLAST results. On reading this Guide, you will Be aBle to: • search a variety of Web-based sequence databases with different query sequences, and alter search parameters; • explain a range of typical search parameters, and the likely impacts on search outputs of changing them; • analyse the information conveyed in search outputs and infer the significance of reported matches; • examine and investigate the annotations of reported matches, and their provenance; and • compare the outputs of different BLAST implementations and evaluate the implications of any differences. finding short words – k-tuples – common to the sequences Being 1 Introduction compared, and using heuristics to join those closest to each other, including the short mis-matched regions Between them. BLAST4 was the second major example of this type of algorithm, From the advent of the first molecular sequence repositories in and rapidly exceeded the popularity of FastA, owing to its efficiency the 1980s, tools for searching dataBases Became essential. DataBase searching is essentially a ‘pairwise alignment’ proBlem, in which the and Built-in statistics. -

Functionfinderspresnotes.Pdf
1 A series of three bases is called a codon which translates into an amino acid. A series of amino acids make a protein. The codon wheel is central to this activity as it translates the DNA sequence into amino acids. To use the wheel you must work from the inside circle out to the outer circle. For example if the first triplet of the sequence is CAT, the amino acid it codes for is Histidine (H). 2 Stress to the students that the amino acid sequence is not the entire amino acid sequence, but a region that we can use to search online databases. Normally, amino acid sequences can be comprised of hundreds of amino acids. By following the link to the Uniprot website at the bottom of the protein profile students will be able to see the full sequence of the protein (if time). 3 4 Before discussing the results, stress to the students that the amino acid sequence is not the entire amino acid sequence, but a region that we can use to search online dbdatabases. Normally, amino acid sequences can be comprised of hundreds of amino acids. By following the link to the Uniprot website at the bottom of the protein profile students will be able to see the full sequence of the protein (if time). The antifreeze protein is essential to prevent organisms like the wolf fish from freezing in icy conditions. Q. Is this just exclusive to this one species / what other species may use antifreeze proteins? Antifreeze proteins are found in other organisms including invertebrates such as the “Snow flea” or springtail, as well as species of plant and some bacteria. -
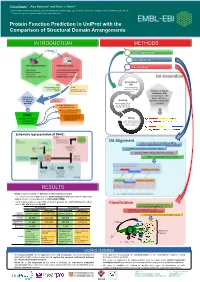
Protein Function Prediction in Uniprot with the Comparison of Structural Domain Arrangements
Tunca Dogan1,*, Alex Bateman1 and Maria J. Martin1 1 European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK * To whom correspondence should be addressed: [email protected] Protein Function Prediction in UniProt with the Comparison of Structural Domain Arrangements INTRODUCTION METHODS Data Preparation DA alignment Classification DA Generation Generation of SAAS DAs (Automatic decision tree- UniRule using InterProScan based rule-generating (Manually curated rules results on UniProtKB system) created by curation team) proteins Definition of domain architecture (DA): concatenation of the Requirement of InterPro IDs of the new approaches Annotations from Grouping for automatic other sources domains on the protein annotation proteins under shared sequence Treats domain hits as DAs & separation of strings (instead of a.a.) learning and test sets Pairwise alignment of DAs for similarity detection DAAC Supervised classification (Domain Architecture of query sequences into Mining functional classes Alignment and functional annotations Classification) from source databases (for learning set) Schematic representation of DAAC: DA Alignment A modified version of Needleman-Wunsch global sequence alignment algorithm to compare the DAs by: - treating the domains as the strings in a sequence - working for 7497 InterPro domains instead of 20 a.a - fast due to reduced total number of operations RESULTS • DAAC system is trained for GO term and EC number prediction -

Uniprot.Ws: R Interface to Uniprot Web Services
Package ‘UniProt.ws’ September 26, 2021 Type Package Title R Interface to UniProt Web Services Version 2.33.0 Depends methods, utils, RSQLite, RCurl, BiocGenerics (>= 0.13.8) Imports AnnotationDbi, BiocFileCache, rappdirs Suggests RUnit, BiocStyle, knitr Description A collection of functions for retrieving, processing and repackaging the UniProt web services. Collate AllGenerics.R AllClasses.R getFunctions.R methods-select.R utilities.R License Artistic License 2.0 biocViews Annotation, Infrastructure, GO, KEGG, BioCarta VignetteBuilder knitr LazyLoad yes git_url https://git.bioconductor.org/packages/UniProt.ws git_branch master git_last_commit 5062003 git_last_commit_date 2021-05-19 Date/Publication 2021-09-26 Author Marc Carlson [aut], Csaba Ortutay [ctb], Bioconductor Package Maintainer [aut, cre] Maintainer Bioconductor Package Maintainer <[email protected]> R topics documented: UniProt.ws-objects . .2 UNIPROTKB . .4 utilities . .8 Index 11 1 2 UniProt.ws-objects UniProt.ws-objects UniProt.ws objects and their related methods and functions Description UniProt.ws is the base class for interacting with the Uniprot web services from Bioconductor. In much the same way as an AnnotationDb object allows acces to select for many other annotation packages, UniProt.ws is meant to allow usage of select methods and other supporting methods to enable the easy extraction of data from the Uniprot web services. select, columns and keys are used together to extract data via an UniProt.ws object. columns shows which kinds of data can be returned for the UniProt.ws object. keytypes allows the user to discover which keytypes can be passed in to select or keys via the keytype argument. keys returns keys for the database contained in the UniProt.ws object . -

Three-Dimensional Structures of Carbohydrates and Where to Find Them
International Journal of Molecular Sciences Review Three-Dimensional Structures of Carbohydrates and Where to Find Them Sofya I. Scherbinina 1,2,* and Philip V. Toukach 1,* 1 N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Science, Leninsky prospect 47, 119991 Moscow, Russia 2 Higher Chemical College, D. Mendeleev University of Chemical Technology of Russia, Miusskaya Square 9, 125047 Moscow, Russia * Correspondence: [email protected] (S.I.S.); [email protected] (P.V.T.) Received: 26 September 2020; Accepted: 16 October 2020; Published: 18 October 2020 Abstract: Analysis and systematization of accumulated data on carbohydrate structural diversity is a subject of great interest for structural glycobiology. Despite being a challenging task, development of computational methods for efficient treatment and management of spatial (3D) structural features of carbohydrates breaks new ground in modern glycoscience. This review is dedicated to approaches of chemo- and glyco-informatics towards 3D structural data generation, deposition and processing in regard to carbohydrates and their derivatives. Databases, molecular modeling and experimental data validation services, and structure visualization facilities developed for last five years are reviewed. Keywords: carbohydrate; spatial structure; model build; database; web-tool; glycoinformatics; structure validation; PDB glycans; structure visualization; molecular modeling 1. Introduction Knowledge of carbohydrate spatial (3D) structure is crucial for investigation of glycoconjugate biological activity [1,2], vaccine development [3,4], estimation of ligand-receptor interaction energy [5–7] studies of conformational mobility of macromolecules [8], drug design [9], studies of cell wall construction aspects [10], glycosylation processes [11], and many other aspects of carbohydrate chemistry and biology. Therefore, providing information support for carbohydrate 3D structure is vital for the development of modern glycomics and glycoproteomics. -

The Role of Uniprot's Protein Sequence Databases in Biomedical Research
Andrew Nightingale1, Tunca Dogan1, Diego Poggioli1, Maria Martin1 and the UniProt Consortium1,2,3 1 EMBL-European Bioinformatics Institute, Cambridge, UK 2 SIB Swiss Institute of Bioinformatics, Geneva, Switzerland 3 Protein Information Resource, Georgetown University, Washington DC & University od Delaware, USA The Role of UniProt's Protein Sequence Databases in Biomedical Research Introduction Mapping diseases to InterPro UniProt provides human disease information with extensive Domains, Variants and ChEMBL cross-references to disease relevant databases such as: Medical Subject Headings (MeSH)1 and Online Mendelian Inheritance in Man Compounds (OMIM)2, Figure 1. In order to further enhance the functional annotations relevance to biomedical research; UniProt has recently developed a pipeline for importing protein altering variants from globally ● 4,246 diseases mapped to 2,337 InterPro domains. recognised genetic variant repositories with the aim to extend the ● 316 InterPro domains from 510 protein entries matched to 3,601 ChEMBL manually curated set of natural protein altering variants provided by ligands. UniProt. By combining these resources UniProt has become a more relevant resource for biomedical research and drug target identification. ● Somatic variants have been found within the binding pockets of proteins Here we describe how users of UniProt can develop methodologies to associated to specific cancer types. utilise the described cross-references, protein structure and functional annotations to explore how structural, functional and chemical ligand annotations can be utilised to identify relationships between a protein and disease causing variants. Mitogen-activated Protein Kinase 4 Example Figure 1: Disease and natural variant annotation for UniProtKB/SwissProt entry for Human BRCA1 Figure 3: MAPK4 binding pocket with analogue inhibitor bound and p.Ser233Ala variant. -

Biogrid Australia (Formerly Biogrid)
BioGrid Australia (formerly BioGrid) Record Linkage The Vision – remove the ‘silos’ Population data Hospital data Research Research Disease Sub‐specialty /research data Project Project Gene Expression 2 1 Protein Expression Genotypes Public Domain Data A generic informatics platform which provides opportunities for collaboration across organisations and expansion to other research areas. Why link databases? • Record linkage for Clinical purposes: – Instant sharing of information across treatment centres (hospitals, GP’s, etc) • Research power: – Increase the sample size – Increase the potential for research collaborations • Main Issues: – Lack of a common identifier at national or even state level (unlike many countries) • Key Question for Linkage: – What level of accuracy is acceptable for each type of linkage? BioGrid ‐ Privacy and Ethics: Required approval from all sites to use their identifying information for linkage purposes . This includes the information being copied to a central location so it can be “matched” against patients from other sites Required a legal opinion that we could use the last 5 digits of the Medicare number as part of the linkage algorithm. There is a provision in the Medicare Act that the whole number cannot be used for identification purposes (a legacy of the Australia Card?) Data available in de‐identified form only, and therefore only suitable for research Health data always kept separate from identifying data Institute-specific data loaded Authorised researchers into institute-specific query the Federated -

Development and Analysis of Web Resources Using Glycoinformatics
Development and Analysis of Web Resources using Glycoinformatics September 2016 Yukie Akune Contents 1 Introduction 1 2 Development of RINGS: Resource for INformatics of Glycomes at Soka 10 2.1 Introduction . 10 2.1.1 Glycome informatics . 10 2.1.2 Glycan related Databases associated with RINGS . 12 2.1.3 Algorithms used in RINGS . 14 2.1.4 RINGS architecture . 17 2.2 Materials and Methods . 18 2.2.1 Glycan Score Matrix development . 19 2.2.2 Glycan Kernel Tool . 23 2.2.3 RINGS Utilities . 25 2.3 Results . 28 2.3.1 Glycan Score Matrices . 28 2.3.2 Glycan Kernel Tool . 30 2.3.3 RINGS Utilities . 32 2.4 Discussions . 38 ii 3 Construction of theoretical N -glycan database 40 3.1 Introduction . 40 3.1.1 Mathematical models for predicting glycan synthetic pathway . 40 3.1.2 A tool for predicting glycan synthetic pathway . 41 3.1.3 glycan related databases associated with theoretical N -glycan database 43 3.1.4 Development of the novel database for glycan synthetic pathway prediction . 46 3.2 Materials and Methods . 47 3.2.1 Glycosyltransferase catalog . 47 3.2.2 Development for a tool for glycosyltransferase candidates prediction 48 3.2.3 Theoretical N -glycan calculation algorithm . 49 3.3 Results . 52 3.3.1 A tool for glycosyltransferase candidates prediction . 52 3.3.2 Glycosyltransferase catalog . 52 3.3.3 Theoretical N -glycan database . 60 3.4 Discussions . 61 4 Conclusion 70 Acknowledgements 72 Bibliography 73 List of Tables 82 List of Figures 84 iii A Source codes i B Glycosyltransferase Catalog iv iv Chapter 1 Introduction Glycans are macromolecular substances that are known to be extremely vital in regards to recognition signals for biological phenomena such as cell-cell communication, biological development and cancer metastases (Fig. -

Introduction to Bioinformatics
INTRODUCTION TO BIOINFORMATICS > Please take the initial BIOINF525 questionnaire: < http://tinyurl.com/bioinf525-questions Barry Grant University of Michigan www.thegrantlab.org BIOINF 525 http://bioboot.github.io/bioinf525_w16/ 12-Jan-2016 Barry Grant, Ph.D. [email protected] Ryan Mills, Ph.D. [email protected] Hongyang Li (GSI) [email protected] COURSE LOGISTICS Lectures: Tuesdays 2:30-4:00 PM Rm. 2062 Palmer Commons Labs: Session I: Thursdays 2:30 - 4:00 PM Session II: Fridays 10:30 - 12:00 PM Rm. 2036 Palmer Commons Website: http://tinyurl.com/bioinf525-w16 Lecture, lab and background reading material plus homework and course announcements MODULE OVERVIEW Objective: Provide an introduction to the practice of bioinformatics as well as a practical guide to using common bioinformatics databases and algorithms 1.1. ‣ Introduction to Bioinformatics 1.2. ‣ Sequence Alignment and Database Searching 1.3 ‣ Structural Bioinformatics 1.4 ‣ Genome Informatics: High Throughput Sequencing Applications and Analytical Methods TODAYS MENU Overview of bioinformatics • The what, why and how of bioinformatics? • Major bioinformatics research areas. • Skepticism and common problems with bioinformatics. Bioinformatics databases and associated tools • Primary, secondary and composite databases. • Nucleotide sequence databases (GenBank & RefSeq). • Protein sequence database (UniProt). • Composite databases (PFAM & OMIM). Database usage vignette • Searching with ENTREZ and BLAST. • Reference slides and handout on major databases. HOMEWORK Complete the initial