High-Throughput Inference of Pairwise Coalescence Times Identifies Signals of Selection and Enriched Disease Heritability
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Investigating the Genetic Basis of Cisplatin-Induced Ototoxicity in Adult South African Patients
--------------------------------------------------------------------------- Investigating the genetic basis of cisplatin-induced ototoxicity in adult South African patients --------------------------------------------------------------------------- by Timothy Francis Spracklen SPRTIM002 SUBMITTED TO THE UNIVERSITY OF CAPE TOWN In fulfilment of the requirements for the degree MSc(Med) Faculty of Health Sciences UNIVERSITY OF CAPE TOWN University18 December of Cape 2015 Town Supervisor: Prof. Rajkumar S Ramesar Co-supervisor: Ms A Alvera Vorster Division of Human Genetics, Department of Pathology, University of Cape Town 1 The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgement of the source. The thesis is to be used for private study or non- commercial research purposes only. Published by the University of Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University of Cape Town Declaration I, Timothy Spracklen, hereby declare that the work on which this dissertation/thesis is based is my original work (except where acknowledgements indicate otherwise) and that neither the whole work nor any part of it has been, is being, or is to be submitted for another degree in this or any other university. I empower the university to reproduce for the purpose of research either the whole or any portion of the contents in any manner whatsoever. Signature: Date: 18 December 2015 ' 2 Contents Abbreviations ………………………………………………………………………………….. 1 List of figures …………………………………………………………………………………... 6 List of tables ………………………………………………………………………………….... 7 Abstract ………………………………………………………………………………………… 10 1. Introduction …………………………………………………………………………………. 11 1.1 Cancer …………………………………………………………………………….. 11 1.2 Adverse drug reactions ………………………………………………………….. 12 1.3 Cisplatin …………………………………………………………………………… 12 1.3.1 Cisplatin’s mechanism of action ……………………………………………… 13 1.3.2 Adverse reactions to cisplatin therapy ………………………………………. -

Copy Number Variation in Fetal Alcohol Spectrum Disorder
Biochemistry and Cell Biology Copy number variation in fetal alcohol spectrum disorder Journal: Biochemistry and Cell Biology Manuscript ID bcb-2017-0241.R1 Manuscript Type: Article Date Submitted by the Author: 09-Nov-2017 Complete List of Authors: Zarrei, Mehdi; The Centre for Applied Genomics Hicks, Geoffrey G.; University of Manitoba College of Medicine, Regenerative Medicine Reynolds, James N.; Queen's University School of Medicine, Biomedical and Molecular SciencesDraft Thiruvahindrapuram, Bhooma; The Centre for Applied Genomics Engchuan, Worrawat; Hospital for Sick Children SickKids Learning Institute Pind, Molly; University of Manitoba College of Medicine, Regenerative Medicine Lamoureux, Sylvia; The Centre for Applied Genomics Wei, John; The Centre for Applied Genomics Wang, Zhouzhi; The Centre for Applied Genomics Marshall, Christian R.; The Centre for Applied Genomics Wintle, Richard; The Centre for Applied Genomics Chudley, Albert; University of Manitoba Scherer, Stephen W.; The Centre for Applied Genomics Is the invited manuscript for consideration in a Special Fetal Alcohol Spectrum Disorder Issue? : Keyword: Fetal alcohol spectrum disorder, FASD, copy number variations, CNV https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 354 Biochemistry and Cell Biology 1 Copy number variation in fetal alcohol spectrum disorder 2 Mehdi Zarrei,a Geoffrey G. Hicks,b James N. Reynolds,c,d Bhooma Thiruvahindrapuram,a 3 Worrawat Engchuan,a Molly Pind,b Sylvia Lamoureux,a John Wei,a Zhouzhi Wang,a Christian R. 4 Marshall,a Richard F. Wintle,a Albert E. Chudleye,f and Stephen W. Scherer,a,g 5 aThe Centre for Applied Genomics and Program in Genetics and Genome Biology, The Hospital 6 for Sick Children, Toronto, Ontario, Canada 7 bRegenerative Medicine Program, University of Manitoba, Winnipeg, Canada 8 cCentre for Neuroscience Studies, Queen's University, Kingston, Ontario, Canada. -

The Genetic Basis of Dupuytren's Disease Gloria Sue Yale School of Medicine, [email protected]
Yale University EliScholar – A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2014 The Genetic Basis Of Dupuytren's Disease Gloria Sue Yale School of Medicine, [email protected] Follow this and additional works at: http://elischolar.library.yale.edu/ymtdl Recommended Citation Sue, Gloria, "The Genetic Basis Of Dupuytren's Disease" (2014). Yale Medicine Thesis Digital Library. 1926. http://elischolar.library.yale.edu/ymtdl/1926 This Open Access Thesis is brought to you for free and open access by the School of Medicine at EliScholar – A Digital Platform for Scholarly Publishing at Yale. It has been accepted for inclusion in Yale Medicine Thesis Digital Library by an authorized administrator of EliScholar – A Digital Platform for Scholarly Publishing at Yale. For more information, please contact [email protected]. The Genetic Basis of Dupuytren’s Disease A Thesis Submitted to the Yale University School of Medicine In Partial Fulfillment of the Requirements for the Degree of Doctor of Medicine by Gloria R. Sue 2014 THE GENETIC BASIS OF DUPUYTREN’S DISEASE. Gloria R. Sue, Deepak Narayan. Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT. Dupuytren’s disease is a common heritable connective tissue disorder of poorly understood etiology. It is thought that oxidative stress pathways may play a critical role in the development of Dupuytren’s disease, given the various disease associations that have been observed. We sought to sequence the mitochondrial and nuclear genomes of patients affected with Dupuytren’s disease using next-generation sequencing technology to potentially identify genes of potential pathogenetic interest. -

Olfactory Receptors in Non-Chemosensory Organs: the Nervous System in Health and Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Sissa Digital Library REVIEW published: 05 July 2016 doi: 10.3389/fnagi.2016.00163 Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease Isidro Ferrer 1,2,3*, Paula Garcia-Esparcia 1,2,3, Margarita Carmona 1,2,3, Eva Carro 2,4, Eleonora Aronica 5, Gabor G. Kovacs 6, Alice Grison 7 and Stefano Gustincich 7 1 Institute of Neuropathology, Bellvitge University Hospital, Hospitalet de Llobregat, University of Barcelona, Barcelona, Spain, 2 Center for Biomedical Research in Neurodegenerative Diseases (CIBERNED), Madrid, Spain, 3 Bellvitge Biomedical Research Institute (IDIBELL), Hospitalet de Llobregat, Barcelona, Spain, 4 Neuroscience Group, Research Institute Hospital, Madrid, Spain, 5 Department of Neuropathology, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands, 6 Institute of Neurology, Medical University of Vienna, Vienna, Austria, 7 Scuola Internazionale Superiore di Studi Avanzati (SISSA), Area of Neuroscience, Trieste, Italy Olfactory receptors (ORs) and down-stream functional signaling molecules adenylyl cyclase 3 (AC3), olfactory G protein a subunit (Gaolf), OR transporters receptor transporter proteins 1 and 2 (RTP1 and RTP2), receptor expression enhancing protein 1 (REEP1), and UDP-glucuronosyltransferases (UGTs) are expressed in neurons of the human and murine central nervous system (CNS). In vitro studies have shown that these receptors react to external stimuli and therefore are equipped to be functional. However, ORs are not directly related to the detection of odors. Several molecules delivered from the blood, cerebrospinal fluid, neighboring local neurons and glial cells, distant cells through the extracellular space, and the cells’ own self-regulating internal homeostasis Edited by: Filippo Tempia, can be postulated as possible ligands. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Deep Sequencing of the Human Retinae Reveals the Expression of Odorant Receptors
fncel-11-00003 January 20, 2017 Time: 14:24 # 1 CORE Metadata, citation and similar papers at core.ac.uk Provided by Frontiers - Publisher Connector ORIGINAL RESEARCH published: 24 January 2017 doi: 10.3389/fncel.2017.00003 Deep Sequencing of the Human Retinae Reveals the Expression of Odorant Receptors Nikolina Jovancevic1*, Kirsten A. Wunderlich2, Claudia Haering1, Caroline Flegel1, Désirée Maßberg1, Markus Weinrich1, Lea Weber1, Lars Tebbe2, Anselm Kampik3, Günter Gisselmann1, Uwe Wolfrum2, Hanns Hatt1† and Lian Gelis1† 1 Department of Cell Physiology, Ruhr-University Bochum, Bochum, Germany, 2 Department of Cell and Matrix Biology, Johannes Gutenberg University of Mainz, Mainz, Germany, 3 Department of Ophthalmology, Ludwig Maximilian University of Munich, Munich, Germany Several studies have demonstrated that the expression of odorant receptors (ORs) occurs in various tissues. These findings have served as a basis for functional studies that demonstrate the potential of ORs as drug targets for a clinical application. To the best of our knowledge, this report describes the first evaluation of the mRNA expression of ORs and the localization of OR proteins in the human retina that set a Edited by: stage for subsequent functional analyses. RNA-Sequencing datasets of three individual Hansen Wang, University of Toronto, Canada neural retinae were generated using Next-generation sequencing and were compared Reviewed by: to previously published but reanalyzed datasets of the peripheral and the macular Ewald Grosse-Wilde, human retina and to reference tissues. The protein localization of several ORs was Max Planck Institute for Chemical Ecology (MPG), Germany investigated by immunohistochemistry. The transcriptome analyses detected an average Takaaki Sato, of 14 OR transcripts in the neural retina, of which OR6B3 is one of the most highly National Institute of Advanced expressed ORs. -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -
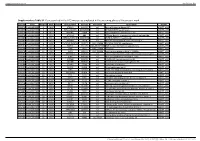
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Chromophobe Renal Cell Carcinoma with and Without Sarcomatoid Change: a Clinicopathological, Comparative Genomic Hybridization, and Whole-Exome Sequencing Study
Am J Transl Res 2015;7(11):2482-2499 www.ajtr.org /ISSN:1943-8141/AJTR0014993 Original Article Chromophobe renal cell carcinoma with and without sarcomatoid change: a clinicopathological, comparative genomic hybridization, and whole-exome sequencing study Yuan Ren1*, Kunpeng Liu1*, Xueling Kang3, Lijuan Pang1, Yan Qi1, Zhenyan Hu1, Wei Jia1, Haijun Zhang1, Li Li1, Jianming Hu1, Weihua Liang1, Jin Zhao1, Hong Zou1*, Xianglin Yuan2, Feng Li1* 1Department of Pathology, School of Medicine, First Affiliated Hospital, Shihezi University, Key Laboratory of Xinjiang Endemic and Ethnic Diseases, Ministry of Education of China, Shihezi, China; 2Tongji Hospital Cancer Center, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China; 3Department of Pathology, Shanghai General Hospital, Shanghai, China. *Equal contributors. Received August 24, 2015; Accepted October 13, 2015; Epub November 15, 2015; Published November 30, 2015 Abstract: Chromophobe renal cell carcinomas (CRCC) with and without sarcomatoid change have different out- comes; however, fewstudies have compared their genetic profiles. Therefore, we identified the genomic alteration- sin CRCC common type (CRCC C) (n=8) and CRCC with sarcomatoid change (CRCC S) (n=4) using comparative genomic hybridization (CGH) and whole-exome sequencing. The CGH profiles showed that the CRCC C group had more chromosomal losses (72 vs. 18) but fewer chromosomal gains (23 vs. 57) than the CRCC S group. Losses of chromosomes 1p, 8p21-23, 10p16-20, 10p12-ter, 13p20-30, and 17p13 and gains of chromosomes 1q11, 1q21-23, 1p13-15, 2p23-24, and 3p21-ter differed between the groups. Whole-exome sequencing showed that the mutational status of 270 genes differed between CRCC (n=12) and normal renal tissues (n=18).