Chem365 Labbook-2011
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

New Era NMR Supplies and Accessories Catalog
analysco NMR Sample Tubes and Accessories from: Over the past twenty years, New Era has been providing the highest quality NMR sample tubes and accessories worldwide, keeping pace with new applications by offering alternative sampling techniques such as capillaries for metabolic samples and apparatus for RDC sample preparation and measurements. In addition, New Era has added a number of other new products to help make sample preparation and experiments easier and more efficient. You can look to New Era for innovation in sampling techniques. Analysco Ltd, 11 Woodlands Close, Milton under Wychwood, Chipping Norton, OX7 6LS, UK T / F: +44 (0)1993 832907 E: [email protected] W: www.analysco.co.uk CONTENTS Page No. Capillaries / Adapters / Support Rods. 21 Page No. Cleaning Brush for sample tubes . 25 NMR Sample Tubes, 3mm, 5mm . 4-7 Cleaning of sample tubes . 32 (including Quartz - 5mm) . 6 Coaxial Insert Cells. 19 NMR Sample Tubes, 8mm to 27mm . 8-9 Contact / Ordering Information . 2 (including Quartz - 10mm) . 8 Cross-Reference for Products. 29-31 NMR Tube Pressure / Volume Data . 32 NMR Tube Specifications . 32 Controlled Atmosphere Valve Sample Tubes . 17 Non-Glass Poly Sample Cells and Accessories Dewars for NMR Applications . 27 (Boron, Fluorine and Silicon studies) . 18 Distributors . 28 pH Electrodes and Solutions. 23 EPR (ESR) Sample Tubes (Quartz) . 19 Pipets / Rubber Bulbs . 22 Pressure Valve Sample Tubes . 17 Gel Sample Tubes (including Presses). 14-15 Probe Inserts (Quartz) . 26 Hazardous Sample Tube System . 16 Raman Sample Tubes . 19 Holders (Racks) for sample tubes . 22 Sample Tube Caps (including Teflon) . 20 Labels (Clear) for sample tubes . -

Equipment Detailsr07
Lab Equipment Details Lab Equipment Glass Flasks 150ml 250ml 500ml Lab Equipment Glass Beakers 150ml 250ml 500ml Lab Equipment Glassware But once removed, only the cap stays highlighted. Droppers critical to Lab An activated Dropper coursework can be found highlights the entire bottle. already in the workspace. Dropper Dropper Dropper Activated In Use Lab Equipment Gastight Syringe Small A pre-filled gastight syringe can be used with a NMR tube to safely fill through the top in preparation for use with the NMR spectrometer. Gastight Syringe NMR Tube with Holder with Holder Lab Equipment NMR Tube Spinner An NMR Tube filled with gas for use with the NMR Spectrometer Simply use the NMR Tube with needs to be inserted into the the Tube Spinner. Spinner before it can be used. NMR Tube NMR Tube Spinner NMR Tube Spinner with Holder with NMR Tube inserted Once the Tube slotted into the holder is inserted into the top part of the spectrometer, users can type in a number for Lab Equipment the frequency they’d like to scan. NMR Spectrometer XL The NMR Tube holder can then be slotted into the highlighted tube on the NMR Spectrometer. NMR Tube filled with appropriate substance is slotted (used with) in the holder. Lift will perpare the Spectrometer for the tube holder insertion. Scan No. Allows the user to change the frequency at which the tube is scanned The NMR Tube holder can then be slotted into the top tube on Lab Equipment the NMR Spectrometer. NMR Spectrometer XL NMR Tube filled with appropriate substance is slotted (used with) in the holder. -

Auxiliary Lab Manual Chem 465L Biochemistry II Lab Spring 2019
Auxiliary Lab Manual Chem 465L Biochemistry II Lab Spring 2019 1 Table of Contents Lab Safety 3 Report Sheet for Experiment 7a 8 A 31P NMR study of a Kinase Reaction 9 Overview of in Vitro and in Vivo 13C Yeast Experiments 12 In Vitro 13C Analysis of Yeast Metabolism 13 In Vivo 13C Analysis of Yeast Metabolism 18 Report Sheet for 13C Analysis of Yeast Metabolism 21 A Reminder of Kinetics Principles 22 NMR Analysis of the Fumarase Reaction 23 DNA Denaturation Lab I - UV 30 DNA Denaturation Lab II - IR 34 DNA Supercoiling Lab 41 DNA/RNA sequence analysis - Computer Lab 45 2 Principles for Safety in the Chemical Laboratory Safe practices in the chemical laboratory are of prime importance. A student should consider it an essential part of his or her educational experience to develop safe and efficient methods of operation in a lab. To do this, one must acquire a basic knowledge of properties of materials present in the lab, and one should realize the types of hazards that exist and the accidents and injuries that can result from ignorance or irresponsibility on the part of the student or a neighbor. Regulations 1. Wear safety goggles at all times while in the laboratory. 2. Report all accidents to the instructor or lab assistant immediately. 3. NEVER eat, drink, chew, or smoke in the laboratory. 4. NEVER leave an experiment unattended. Inform the lab assistant if you must leave the lab. 5. After the experiment is completed, turn all equipment off, making sure it is properly stored, and clean your area. -

NMR Facility User Guide
NMR Facility User Guide Analytical Instrumentation Center School of Pharmacy University of Wisconsin–Madison F2 (ppm) 1.5 2.0 2.5 3.0 3.5 4.0 4.5 75 70 65 60 55 50 45 40 35 30 F1 (ppm) Thomas C. Stringfellow Sixth Edition Version 2019-05-01 Table of Contents 1 Introduction 1 1.1 NMR Spectrometers .................................... 1 1.1.1 AV-400 Spectrometer ................................ 2 1.1.2 UI-500 Spectrometer ................................ 2 1.2 Supporting Computers and Software ............................ 5 1.3 NMR Facility Policies ................................... 6 1.3.1 Access and Use ................................... 6 1.3.2 User Training .................................... 6 1.3.3 Fees and Services .................................. 6 1.3.4 Reserving Instrument Time ............................. 6 1.3.5 Laboratory Safety and Health Issues ........................ 7 1.3.6 AV-400 Sample-Management Policies ....................... 9 1.3.7 Incident Report Form ................................ 10 2 Computers and Software 12 2.1 Information for Bruker Users ................................ 12 2.1.1 IconNMR Web View ................................ 13 2.2 Information for Varian Users ................................ 13 2.2.1 VNMR 6.1C Users Take Note! ........................... 13 2.2.2 Sun Desktop Environments: CDE versus OWD .................. 14 2.3 NMR Facility Computer Network ............................. 15 2.4 Samba Server Connections to NMR Facility Disk Shares ................. 15 2.4.1 Connection to Samba Shares from Microsoft Windows Clients .......... 16 2.4.2 Connection to Samba Shares from Mac OS X Clients ............... 17 2.5 X-Win32 Connectivity to Sun Computers ......................... 17 2.5.1 Installing X-Win32 ................................. 18 2.5.2 Configuring X-Win32 ............................... 19 2.5.3 Configuring the PC Monitor Resolution ..................... -

Secure Series NMR Sample Tubes
in the past 50 years ...with help from Norell you have made new discoveries through cutting edge research creating a new world of possibilities with life- changing breakthroughs. Here’s to another 50 years with Norell by your side. Table of Contents Secure Series NMR Tubes Accessories Secure Series/NorLoc II 4 pH Electrodes & Cables for NMR Tubes 46 Secure 55 Series 5mm 5 Pasteur Pipettes 47 Secure 33 Series 5mm & 3mm 6, 7 Optimizer Inserts™ for 5mm Turbines 48, 49 Toroids 50 Select Series NMR Tubes Spinner Brushes 50 10mm Select Series 8 Fluoropolymer NMR Tube Liners 51 5mm Select Series 8 Fluoropolymer Liner Tube Kits 51 3mm & 4mm Select Series 9 3mm & 5mm NMR Tube Brush 51 5mm & 3mm NMR Sample Tubes for Ceramic Turbines 10, 11 72 Position NMR Tube Rack 52 5mm NMR Tube Carriers 52 Standard Series NMR Tubes Fluoropolymer NMR Tube Caps 52 5mm Standard Series 12, 13 NMR Tube Cleaner, 5 Position 53 10mm Standard Series 14, 15 NRS-250 Surfactant 53 Tube Washing Unit 53 Special Purpose Tubes NorLoc™ Generation II 5mm & 3mm Caps 4, 54, 55, 56, 57 Sample Vault™ NMR Tubes & Caps 16, 17, 18, 19 Economy NMR Tube Caps 58 Closures for Open Port Caps 18, 19 PTFE Syringe Tubing 59 Quartz NMR Tubes 20, 21 Cuvettes 60 Quartz EPR Tubes 21 Fluoropolymer Column Packing 60 Valved NMR Tubes 22, 23 Coaxial Inserts for NMR Tubes 61 Valved NMR Tubes for Intermediate Pressure 24, 25, 26, 27 Polypropylene Syringes 61 Valved NMR Tubes for High Pressure 28, 29, 30, 31 5mm & 10mm NMR Tube Septa 62 Screw-Cap NMR Tubes 32 Bruker Match™ NMR Tube Rack 62 Screw Cap NMR -

ACE GLASS INCORPORATED PRICE LIST 2021 Effective January 1, 2021
Custom Amber Coating ACE GLASS INCORPORATED PRICE LIST 2021 Effective January 1, 2021 Contact your local sales rep today for more information 1-800-223-4524 | www.aceglass.com Order by Code ACE Glassware Discounts Each item in this catalog has a two or three-digit code in addition to All ACE-manufactured glassware, identified with a spade (♠), listed the four or five-digit number. No other ordering information is needed in this catalog is subject to the following dollar value discounts. since each individual size, capacity, etc. has its own code. Example: Items marked by a star (★) or that have no designation whatsoever 5000 10/30 top 14/35 bottom would be ordered as 5000-05. are not subject to this discount. The majority of items listed in this catalog are normally available 10% on purchases of $500.00 and over from stock at our plant in Vineland, NJ. 12% on purchases of $1000.00 and over In the event your entire order cannot be filled immediately, a partial 15% on purchases of $1500.00 and over shipment will be sent, with the back-ordered items following shortly. Terms: Net 30 days (Domestic only) If you should desire the entire order to be sent in one shipment, Minimum Order: $25.00 please specify on your purchase order. Unless otherwise specified on the order, we will ship material by what we consider the “best way.” All quantities in this catalog are “each” unless otherwise noted. Ways to Order Contact us for current pricing or visit www.aceglass.com Ace Glass products are also available from our many lab distribution partners, particularly VWR International and Sigma Aldrich. -

Technique Notes
Dros. Inf. Serv. 90 (2007) 131 Technique Notes Methods and rationale for high-resolution magnetic resonance imaging (MRI) of Drosophila, using an 18.8 Tesla NMR spectrometer. Null, Brian1*, Corey W. Liu2, Maj Hedehus3, Steven Conolly4, and Ronald W. Davis1. 1Stanford Genome Technology Ctr/Bio-X Program; 2Stanford Magnetic Resonance Laboratory; 3Varian Inc, NMR Instruments; 4U.C. Berkeley, Dept. of Bioengineering; *Corresponding author Introduction Magnetic resonance imaging (MRI) is proven as an important tool for the study of thick or opaque tissues in living organisms, and its application to the study of development and biomedicine in the smallest model organisms is an exciting frontier. NMR spectrometers, while not typically used for imaging, are capable of generating extremely high magnetic fields, up to approximately 20 Tesla, whereas the more familiar MRI devices used to image humans in the clinical setting operate at only about one Tesla. High field strength is especially important for tiny specimens like the fruit fly, to increase signal to noise ratio and NMR spectral resolution for quantitation of metabolites in vivo. The small sample dimensions of the NMR spectrometer are ideal for the study of Drosophila and other small model organisms. Further, the ongoing innovation of MR contrast agents which can act as in vivo indicators of physiological status such as calcium ion concentration and gene expression, combined with the robustness of Drosophila as a model organism with a diverse array of genetic tools and genomic data, would make in vivo imaging and spectroscopy a highly desirable technique for the study of Drosophila. Over the past several decades, tremendous advances have been made in the capabilities of magnetic resonance methods for imaging and spectroscopic measurement in human subjects. -

Alcohol Oxidations – Beyond Labz Virtual Chemlab Activity
Alcohol Oxidations – Beyond Labz Virtual ChemLab Activity Purpose: In this virtual experiment, you will be performing two oxidation reactions of benzyl alcohol, a primary alcohol. Primary alcohols can be oxidized to aldehydes or carboxylic acids, depending on the reagents used. You will be setting up oxidation reactions using chromic acid (H2CrO4) and pyridinium chlorochromate (PCC), and comparing the products of the two reactions. You will be monitoring the reactions using thin-layer chromatography (TLC) and analyzing IR and NMR spectra of the reactants and products. O further O OH oxidation oxidation H OH benzyl alcohol benzaldehyde benzoic acid Figure 1. Oxidation reactions of benzyl alcohol Introduction: Primary alcohols can be oxidized to aldehydes or carboxylic acids, depending on the reagents used. For many years, chromium reagents were commonly used for alcohol oxidations. Because of the toxicity of chromium-based reagents, many safer oxidizing agents have been developed and are more commonly used. As this lab is virtual, we can safely explore the reactivity trends for the older, chromium-based reagents. The two reaction conditions we will be exploring in this virtual experiment are: • Jones oxidation: This reaction uses chromic acid (H2CrO4) as the oxidizing agent. Chromic acid can be formed by dissolving sodium dichromate (Na2Cr2O7) or chromium trioxide (CrO3) in aqueous sulfuric acid (H2SO4). • PCC oxidation: This reaction uses pyridinium chlorochromate (PCC) in an anhydrous solvent, typically dichloromethane (CH2Cl2). The virtual lab does not give us CH2Cl2 as an option for a solvent, so we will substitute diethyl ether (CH3CH2OCH2CH3, or Et2O). Once your two virtual experiments are complete, you will decide which set of conditions oxidizes primary alcohols to aldehydes, and which oxidizes primary alcohols to carboxylic acids. -

NMR Solvents Use and Handling of NMR Solvents
NMR Solvents Use and Handling of NMR Solvents NMR Solvents Use and Handling of NMR Solvents NMR Most deuterated NMR solvents readily absorb moisture. To minimize the chance of water contamination, use carefully dried NMR tubes and handle NMR solvents in a dry atmosphere. How to Obtain a Nearly Moisture-free Surface 1. Dry glassware at ~150 °C for 24 hours and cool under an inert atmosphere. 2. Rinse the NMR tube with the deuterated solvent prior to preparing the sample. This allows for a complete exchange of protons from any residual moisture on the glass surface. 3. For less demanding applications, a nitrogen blanket over the sample preparation setup may be adequate. How to Avoid Sources of Impurities and Chemical Residues 1. Use clean, dry glassware and PTFE accessories. 2. Use a vortex mixer instead of shaking the tube contents. The latter action can introduce contaminants from the NMR tube cap. 3. Residual chemical vapor from equipment can be a source of impurities; residual acetone in pipette bulbs is a common example. How to Remove Solvent Residue 1. Protonated solvent residue can be removed by co-evaporation. 2. Use a small quantity of the desired deuterated solvent, a brief high- vacuum drying (5‑10 min), and then prepare the NMR sample. 3. Solvents such as chloroform-d, benzene-d6, and toluene-d8, also remove residual water azeotropically. How to Avoid TMS Evaporation 1. Extended storage of TMS-containing solvents can lead to some loss of TMS. Storing these solvents in Sure/Seal™ bottles virtually eliminates such a loss.* 2. -

In Situ Ph Determination Based on the NMR Analysis of ¹H-NMR Signal Intensities and ¹⁹F-NMR Chemical Shifts
Scholars' Mine Masters Theses Student Theses and Dissertations Summer 2017 In situ pH determination based on the NMR analysis of ¹H-NMR signal intensities and ¹⁹F-NMR chemical shifts Ming Huang Follow this and additional works at: https://scholarsmine.mst.edu/masters_theses Part of the Physical Chemistry Commons Department: Recommended Citation Huang, Ming, "In situ pH determination based on the NMR analysis of ¹H-NMR signal intensities and ¹⁹F- NMR chemical shifts" (2017). Masters Theses. 7867. https://scholarsmine.mst.edu/masters_theses/7867 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. IN SITU PH DETERMINATION BASED ON THE NMR ANALYSIS OF 1H – NMR SIGNAL INTENSITIES AND 19F – NMR CHEMICAL SHIFTS by MING HUANG A THESIS Presented to the Faculty of the Graduate School of the MISSOURI UNIVERSITY OF SCIENCE AND TECHNOLOGY In Partial Fulfillment of the Requirements for the Degree MASTER OF SCIENCE IN CHEMISTRY 2017 Approved by: Klaus Woelk, Advisor V. Prakash Reddy Paul Ki-souk Nam 2017 Ming Huang All Rights Reserved iii ABSTRACT The pH of an NMR sample can be measured directly by NMR experiments of signal intensities, chemical shifts, or relaxation time constants that depend on the pH. The 1H NMR peak intensities of the pH indicator phenolphthalein change as it changes from the OH-depleted form to the OH-rich form in the range of pH = 11.1 to 12.7. -
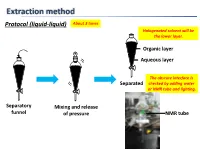
Extraction Method Protocol (Liquid-Liquid) About 3 Times Halogenated Solvent Will Be the Lower Layer
Extraction method Protocol (liquid-liquid) About 3 times Halogenated solvent will be the lower layer. Organic layer Aqueous layer The obscure interface is Separated checked by adding water or NMR tube and lighting. Separatory Mixing and release funnel of pressure NMR tube Extraction method Protocol (liquid-liquid, small scale) Suck organic layer by pipette. Organic layer Stirring bar Aqueous layer Test tube Mixing Separated Tips Vortex mixer is also applicable. 1. To check completion of the extraction, analyze the aqueous layer by TLC. 2. To extract highly polar products, use CH2Cl2:MeOH (9:1) or THF after adding brine to the aqueous layer or evaporate the aqueous layer. 3. To avoid emulsions, gently shake and swirl the funnel. 4. To destroy emulsions, add brine or hexane to the mixture or filter out impurities by Celite. 5. To remove DMSO or DMF, use hexane/EtOAc (3/1) or Et2O. Extraction method 6. To remove less-polar byproducts, firstly dissolve a product to aqueous layer and then to organic layer again by adjusting the pH of aqueous layer. Ex.) Back extraction of carboxylic acid NaOH aq. HCl aq. pH↑ pH↓ Organic layer Organic layer Organic layer (Here!) Aqueous layer Basic Acidic aqueous layer aqueous layer (Here!) Less-polar byproducts are removed. Data of common solvents The organic solvent should 1. Dissolve a product to be extracted. 2. Not react with a product to be extracted. 3. Not react with or be miscible with water. 4. Have a low boiling point. Dehydration method Protocol Drying agent Cotton or Drying agent Then filtered Aspirator can be used. -

Nmr Consumables and Accessories
NMR CONSUMABLES AND ACCESSORIES Features • Control temperature from -90°C to +100°C • Flow rate of up to 2 scfm for solid state NMR samples • ±0.1°C temperature stability • Digital Temperature Control • Choice of multiple non-magnetic delivery line lengths • CE compliant FTS Systems AirJet™ XR sample coolers provide sample temperature control for X-ray diffraction, NMR, EPR, and other applications. These mechanically- refrigerated systems control the AirJet™ XR temperature of a supplied gas stream to between -90°C and +100°C. An optional Sample Cooler air dryer allows for the use of a house- for Liquid- and Solid-State NMR compressed air supply. The unique temperature controller provides precise regulation of heat input to produce a temperature stability of ±0.1°C. The non-magnetic variable length flexible SP Scientific • 3538 Main street, Stone Ridge, NY 12484 USA delivery lines allow you to position Phone: 845-255-5000 • Fax: 845-687-7481 the air stream for proper sample E-mail: [email protected] • www.spscientific.com temperature control. NMR Tube Technical Information Liquid Phase NMR of Contents Table Precision Tubes 6 - 14 Outer Diameter & Inner Diameter Precision Step-Down Tubes 14 Outer Diameter (O.D.) - A measure of the distance across the Precision Quartz/Suprasil Tubes 15 - 16 center of the tube from the outermost surfaces. Economy Tubes 17 Inner Diameter (I.D.) - A measure of the distance across the High Throughput & Sample Jet Tubes 18 center of the tube from the innermost surfaces. NEW Benchtop Spectrometer Tubes 19 Bar Code Tubes 19 Concentricity NEW Reaction Monitoring System 20 A measurement of variation in the radial centers, measured at Double Layered Tubes 20 the inner and outer walls.