Portada Tesis
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

ASSESSING DIAGNOSTIC and THERAPEUTIC TARGETS in OBESITY- ASSOCIATED LIVER DISEASES Noemí Cabré Casares
ASSESSING DIAGNOSTIC AND THERAPEUTIC TARGETS IN OBESITY- ASSOCIATED LIVER DISEASES Noemí Cabré Casares ADVERTIMENT. L'accés als continguts d'aquesta tesi doctoral i la seva utilització ha de respectar els drets de la persona autora. Pot ser utilitzada per a consulta o estudi personal, així com en activitats o materials d'investigació i docència en els termes establerts a l'art. 32 del Text Refós de la Llei de Propietat Intel·lectual (RDL 1/1996). Per altres utilitzacions es requereix l'autorització prèvia i expressa de la persona autora. En qualsevol cas, en la utilització dels seus continguts caldrà indicar de forma clara el nom i cognoms de la persona autora i el títol de la tesi doctoral. No s'autoritza la seva reproducció o altres formes d'explotació efectuades amb finalitats de lucre ni la seva comunicació pública des d'un lloc aliè al servei TDX. Tampoc s'autoritza la presentació del seu contingut en una finestra o marc aliè a TDX (framing). Aquesta reserva de drets afecta tant als continguts de la tesi com als seus resums i índexs. ADVERTENCIA. El acceso a los contenidos de esta tesis doctoral y su utilización debe respetar los derechos de la persona autora. Puede ser utilizada para consulta o estudio personal, así como en actividades o materiales de investigación y docencia en los términos establecidos en el art. 32 del Texto Refundido de la Ley de Propiedad Intelectual (RDL 1/1996). Para otros usos se requiere la autorización previa y expresa de la persona autora. En cualquier caso, en la utilización de sus contenidos se deberá indicar de forma clara el nombre y apellidos de la persona autora y el título de la tesis doctoral. -

C12) United States Patent (IO) Patent No.: US 10,774,349 B2 San Et Al
I1111111111111111 1111111111 111111111111111 IIIII IIIII IIIII IIIIII IIII IIII IIII US010774349B2 c12) United States Patent (IO) Patent No.: US 10,774,349 B2 San et al. (45) Date of Patent: Sep.15,2020 (54) ALPHA OMEGA BIFUNCTIONAL FATTY 9/1029 (2013.01); C12N 9/13 (2013.01); ACIDS C12N 9/16 (2013.01); C12N 9/88 (2013.01); C12N 9/93 (2013.01) (71) Applicant: William Marsh Rice University, (58) Field of Classification Search Houston, TX (US) None See application file for complete search history. (72) Inventors: Ka-Yiu San, Houston, TX (US); Dan Wang, Houston, TX (US) (56) References Cited (73) Assignee: William Marsh Rice University, U.S. PATENT DOCUMENTS Houston, TX (US) 9,994,881 B2 * 6/2018 Gonzalez . Cl2N 9/0006 2016/0090576 Al 3/2016 Garg et al. ( *) Notice: Subject to any disclaimer, the term ofthis patent is extended or adjusted under 35 FOREIGN PATENT DOCUMENTS U.S.C. 154(b) by 152 days. WO W02000075343 12/2000 (21) Appl. No.: 15/572,099 WO W02016179572 11/2016 (22) PCT Filed: May 7, 2016 OTHER PUBLICATIONS (86) PCT No.: PCT/US2016/031386 Choi, K. H., R. J. Heath, and C. 0. Rock. 2000. 13-Ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in § 371 (c)(l), branched-chain fatty acid biosynthesis. J. Bacteriol. 182:365-370. (2) Date: Nov. 6, 2017 He, X., and K. A. Reynolds. 2002. Purification, characterization, and identification of novel inhibitors of the beta-ketoacyl-acyl (87) PCT Pub. No.: WO2016/179572 carrier protein synthase III (FabH) from Staphylococcus aureus. -

In Vivo Dual RNA-Seq Analysis Reveals the Basis for Differential Tissue Tropism of Clinical Isolates of Streptococcus Pneumoniae
In Vivo Dual RNA-Seq Analysis Reveals the Basis for Differential Tissue Tropism of Clinical Isolates of Streptococcus pneumoniae Vikrant Minhas,1,4 Rieza Aprianto,2,4 Lauren J. McAllister,1 Hui Wang,1 Shannon C. David,1 Kimberley T. McLean,1 Iain Comerford,3 Shaun R. McColl,3 James C. Paton,1,5,6,* Jan-Willem Veening,2,5 and Claudia Trappetti,1,5 Supplementary Information Supplementary Table 1. Pneumococcal differential gene expression in the lungs 6 h post-infection, 9-47-Ear vs 9-47M. Genes with fold change (FC) greater than 2 and p < 0.05 are shown. FC values highlighted in blue = upregulated in 9-47-Ear, while values highlighted in red = upregulated in 9- 47M. Locus tag in 9-47- Product padj FC Ear Sp947_chr_00844 Sialidase B 3.08E-10 313.9807 Sp947_chr_02077 hypothetical protein 4.46E-10 306.9412 Sp947_chr_00842 Sodium/glucose cotransporter 2.22E-09 243.4822 Sp947_chr_00841 N-acetylneuraminate lyase 4.53E-09 227.7963 scyllo-inositol 2-dehydrogenase Sp947_chr_00845 (NAD(+)) 4.36E-09 221.051 Sp947_chr_00848 hypothetical protein 1.19E-08 202.7867 V-type sodium ATPase catalytic subunit Sp947_chr_00853 A 1.29E-06 100.5411 Sp947_chr_00846 Beta-glucoside kinase 3.42E-06 98.18951 Sp947_chr_00855 V-type sodium ATPase subunit D 8.34E-06 85.94879 Sp947_chr_00851 V-type sodium ATPase subunit C 2.50E-05 72.46612 Sp947_chr_00843 hypothetical protein 2.17E-05 65.97758 Sp947_chr_00839 HTH-type transcriptional regulator RpiR 3.09E-05 61.28171 Sp947_chr_00854 V-type sodium ATPase subunit B 1.32E-06 50.86992 Sp947_chr_00120 hypothetical protein 3.00E-04 -

Greg's Awesome Thesis
Analysis of alignment error and sitewise constraint in mammalian comparative genomics Gregory Jordan European Bioinformatics Institute University of Cambridge A dissertation submitted for the degree of Doctor of Philosophy November 30, 2011 To my parents, who kept us thinking and playing This dissertation is the result of my own work and includes nothing which is the out- come of work done in collaboration except where specifically indicated in the text and acknowledgements. This dissertation is not substantially the same as any I have submitted for a degree, diploma or other qualification at any other university, and no part has already been, or is currently being submitted for any degree, diploma or other qualification. This dissertation does not exceed the specified length limit of 60,000 words as defined by the Biology Degree Committee. November 30, 2011 Gregory Jordan ii Analysis of alignment error and sitewise constraint in mammalian comparative genomics Summary Gregory Jordan November 30, 2011 Darwin College Insight into the evolution of protein-coding genes can be gained from the use of phylogenetic codon models. Recently sequenced mammalian genomes and powerful analysis methods developed over the past decade provide the potential to globally measure the impact of natural selection on pro- tein sequences at a fine scale. The detection of positive selection in particular is of great interest, with relevance to the study of host-parasite conflicts, immune system evolution and adaptive dif- ferences between species. This thesis examines the performance of methods for detecting positive selection first with a series of simulation experiments, and then with two empirical studies in mammals and primates. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Downloaded from (2007)
ARTICLE https://doi.org/10.1038/s41467-020-14796-x OPEN Alterations in promoter interaction landscape and transcriptional network underlying metabolic adaptation to diet ✉ Yufeng Qin1, Sara A. Grimm2, John D. Roberts1, Kaliopi Chrysovergis1 & Paul A. Wade 1 Metabolic adaptation to nutritional state requires alterations in gene expression in key tis- sues. Here, we investigated chromatin interaction dynamics, as well as alterations in cis- 1234567890():,; regulatory loci and transcriptional network in a mouse model system. Chronic consumption of a diet high in saturated fat, when compared to a diet high in carbohydrate, led to dramatic reprogramming of the liver transcriptional network. Long-range interaction of promoters with distal regulatory loci, monitored by promoter capture Hi-C, was regulated by metabolic status in distinct fashion depending on diet. Adaptation to a lipid-rich diet, mediated largely by nuclear receptors including Hnf4α, relied on activation of preformed enhancer/promoter loops. Adaptation to carbohydrate-rich diet led to activation of preformed loops and to de novo formation of new promoter/enhancer interactions. These results suggest that adapta- tion to nutritional changes and metabolic stress occurs through both de novo and pre-existing chromatin interactions which respond differently to metabolic signals. 1 Eukaryotic Transcriptional Regulation Group, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA. 2 Integrative Bioinformatics -

Suppression of Fatty Acid Oxidation by Thioesterase Superfamily Member
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440732; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Suppression of Fatty Acid Oxidation by Thioesterase Superfamily Member 2 in Skeletal Muscle Promotes Hepatic Steatosis and Insulin Resistance Norihiro Imai1, Hayley T. Nicholls1, Michele Alves-Bezerra1, Yingxia Li1, Anna A. Ivanova2, Eric A. Ortlund2, and David E. Cohen1 1Division of Gastroenterology and Hepatology, Joan & Sanford I. Weill Department of Medicine, Weill Cornell Medical College, NY 10021 USA 2Department of Biochemistry, Emory University, Atlanta, GA 30322 USA Current addresses: Norihiro Imai - Department of Gastroenterology and Hepatology, Nagoya University School of Medicine, Aichi 4668560 Japan Michele Alves-Bezerra - Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030 USA bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440732; this version posted April 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Figure number: 8 Supplemental figure number: 10 Supplemental table number: 2 References: 48 Keywords: Hepatic steatosis, obesity, acyl-CoA thioesterase, fatty acid oxidation, insulin resistance Conflict of interest: The authors have declared that no conflict of interest exists. Author contributions: N.I.: designed research studies, conducted experiments, acquired data, analyzed data and wrote manuscript. H.T.N.: conducted experiments and analyzed data, M.A.B.: designed research studies and conducted experiments, Y.L.: acquired data, A.A.I.: conducted experiments and analyzed data, E.A.O.: analyzed data, D.E.C.: designed research studies, analyzed data and wrote manuscript. -

Structural Basis for Recruitment of Tandem Hotdog Domains in Acyl-Coa Thioesterase 7 and Its Role in Inflammation
Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation Jade K. Forwood*†‡, Anil S. Thakur*, Gregor Guncar*§¶, Mary Marfori*, Dmitri Mouradov*, Weining Meng*§, Jodie Robinson§ʈ, Thomas Huber*, Stuart Kellie*§ʈ, Jennifer L. Martin*§**, David A. Hume*§ʈ**††, and Bostjan Kobe*‡§** *School of Molecular and Microbial Sciences, §Institute for Molecular Bioscience, ʈCooperative Research Centre for Chronic Inflammatory Diseases, and **Australian Research Council Special Research Centre for Functional and Applied Genomics, University of Queensland, Brisbane, Queensland 4072, Australia Edited by Gregory A. Petsko, Brandeis University, Waltham, MA, and approved May 10, 2007 (received for review February 2, 2007) Acyl-CoA thioesterases (Acots) catalyze the hydrolysis of fatty long-chain acyl-CoA substrates with fatty acid chains of 8–16 acyl-CoA to free fatty acid and CoA and thereby regulate lipid carbon atoms (C8–C16) (7, 13). Acot7 contains a pair of fused metabolism and cellular signaling. We present a comprehensive thioesterase domains that share Ϸ30% sequence identity, with each structural and functional characterization of mouse acyl-CoA thio- thioesterase domain predicted to have the hotdog fold structure esterase 7 (Acot7). Whereas prokaryotic homologues possess a with an ␣-helix sausage wrapped by a -sheet bun (14–16). single thioesterase domain, mammalian Acot7 contains a pair of Here, we present the crystal structures of the N- and C- domains in tandem. We determined the crystal structures of both terminal thioesterase domains (N- and C-domains) of mouse the N- and C-terminal domains of the mouse enzyme, and inferred Acot7, refined at 1.8 and 2.4 Å resolution, respectively. -

The Emerging Role of Acyl-Coa Thioesterases and Acyltransferases in Regulating Peroxisomal Lipid Metabolism☆
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1822 (2012) 1397–1410 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Review The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism☆ Mary C. Hunt a,⁎, Marina I. Siponen b,1, Stefan E.H. Alexson c a Dublin Institute of Technology, School of Biological Sciences, College of Sciences & Health, Kevin Street, Dublin 8, Ireland b Department of Medical Biochemistry and Biophysics, Structural Genomics Consortium, Karolinska Institutet, SE-171 77 Stockholm, Sweden c Karolinska Institutet, Department of Laboratory Medicine, Division of Clinical Chemistry, Karolinska University Hospital at Huddinge, SE-141 86 Stockholm, Sweden article info abstract Article history: The importance of peroxisomes in lipid metabolism is now well established and peroxisomes contain approx- Received 15 November 2011 imately 60 enzymes involved in these lipid metabolic pathways. Several acyl-CoA thioesterase enzymes Received in revised form 3 March 2012 (ACOTs) have been identified in peroxisomes that catalyze the hydrolysis of acyl-CoAs (short-, medium-, Accepted 16 March 2012 long- and very long-chain), bile acid-CoAs, and methyl branched-CoAs, to the free fatty acid and coenzyme Available online 23 March 2012 A. A number of acyltransferase enzymes, which are structurally and functionally related to ACOTs, have also been identified in peroxisomes, which conjugate (or amidate) bile acid-CoAs and acyl-CoAs to amino acids, Keywords: Acyl-CoA thioesterase resulting in the production of amidated bile acids and fatty acids. -

Flavone Effects on the Proteome and Transcriptome of Colonocytes in Vitro and in Vivo and Its Relevance for Cancer Prevention and Therapy
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährungsphysiologie Flavone effects on the proteome and transcriptome of colonocytes in vitro and in vivo and its relevance for cancer prevention and therapy Isabel Winkelmann Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. D. Haller Prüfer der Dissertation: 1. Univ.-Prof. Dr. H. Daniel 2. Univ.-Prof. Dr. U. Wenzel (Justus-Liebig-Universität Giessen) 3. Prof. Dr. E.C.M. Mariman (Maastricht University, Niederlande) schriftliche Beurteilung Die Dissertation wurde am 24.08.2009 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 25.11.2009 angenommen. Die Forschung ist immer auf dem Wege, nie am Ziel. (Adolf Pichler) Table of contents 1. Introduction .......................................................................................................... 1 1.1. Cancer and carcinogenesis .................................................................................. 2 1.2. Colorectal Cancer ............................................................................................... 3 1.2.1. Hereditary forms of CRC ........................................................................................ 4 1.2.2. Sporadic forms of CRC .......................................................................................... -

Analysis of the Impact of Population Copy Number Variation in Introns
UNIVERSIDAD AUTÓNOMA DE MADRID Programa de Doctorado en Biociencias Moleculares Analysis of the Impact of Population Copy Number Variation in Introns Maria Rigau de Llobet Madrid, 2019 DEPARTMENT OF BIOCHEMISTRY UNIVERSIDAD AUTÓNOMA DE MADRID FACULTY OF MEDICINE Analysis of the Impact of Population Copy Number Variation in Introns Maria Rigau de Llobet Graduate in Biology Thesis Directors: Dr. Alfonso Valencia Herrera and Dr. Daniel Rico Rodríguez Barcelona Supercomputing Center I Certificado del director o directores Dr Alfonso Valencia Herrera, Head of the Computational Biology Life Sciences Group (Barcelona Supercomputing Center) and Dr Daniel Rico Rodríguez, Head of the Chromatin, Immunity and Bioinformatics Group (Institute of Cellular Medicine, Newcastle, UK) CERTIFY: That Ms. Maria Rigau de Llobet, Graduate in Biology by the University Pompeu Fabra (Barcelona, Spain), has completed her Doctoral Thesis entitled “Analysis of the Impact of Population Copy Number Variation in Introns” under our supervision and meets the necessary requirements to obtain the PhD degree in Molecular Biosciences. To this purpose, she will defend her doctoral thesis at the Universidad Autó noma de Madrid. We hereby authorize its defense in front of the appropriate Thesis evaluation panel. We issue this certificate in Madrid on April 30th 2019 Alfonso Valencia Herrera Daniel Rico Rodríguez Thesis Director Thesis Director III This thesis was supported by a “La Caixa” – Severo Ochoa International PhD Programme V Aknowledgments Para empezar, me gustaría dar las gracias a mis dos directores de tesis, Alfonso Valencia y Daniel Rico. Muchas gracias Alfonso por la oportunidad de hacer el doctorado en tu grupo. Puedo decir sin dudar que ha sido el comienzo de una carrera profesional con la que disfruto. -
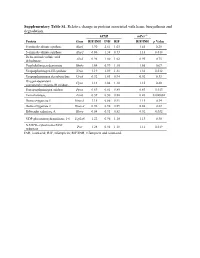
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-