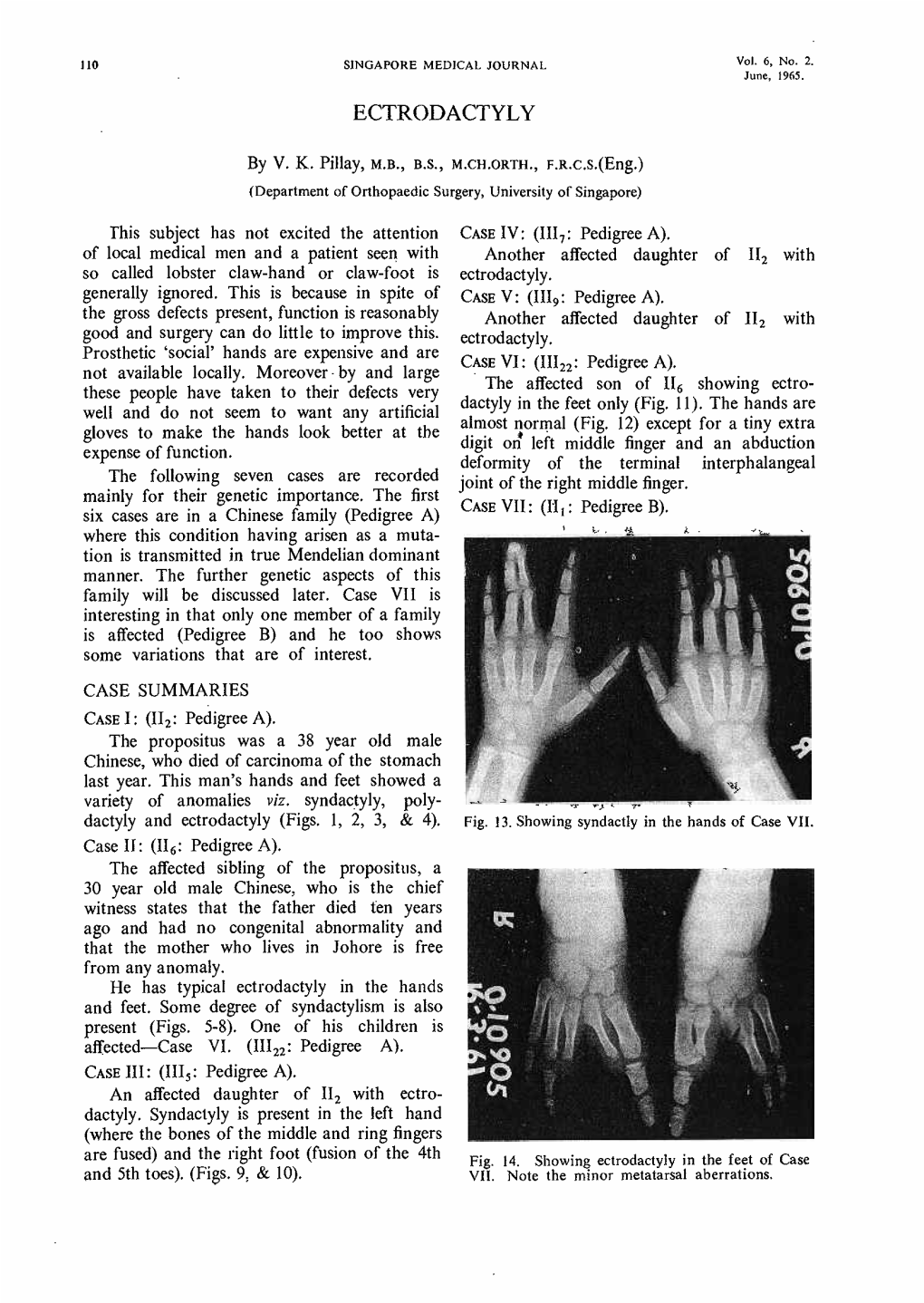
Ectrodactyly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Frontosphenoidal Synostosis: a Rare Cause of Unilateral Anterior Plagiocephaly
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Childs Nerv Syst (2007) 23:1431–1438 DOI 10.1007/s00381-007-0469-4 ORIGINAL PAPER Frontosphenoidal synostosis: a rare cause of unilateral anterior plagiocephaly Sandrine de Ribaupierre & Alain Czorny & Brigitte Pittet & Bertrand Jacques & Benedict Rilliet Received: 30 March 2007 /Published online: 22 September 2007 # Springer-Verlag 2007 Abstract Conclusion Frontosphenoidal synostosis must be searched Introduction When a child walks in the clinic with a in the absence of a coronal synostosis in a child with unilateral frontal flattening, it is usually associated in our anterior unilateral plagiocephaly, and treated surgically. minds with unilateral coronal synostosis. While the latter might be the most common cause of anterior plagiocephaly, Keywords Craniosynostosis . Pediatric neurosurgery. it is not the only one. A patent coronal suture will force us Anterior plagiocephaly to consider other etiologies, such as deformational plagio- cephaly, or synostosis of another suture. To understand the mechanisms underlying this malformation, the development Introduction and growth of the skull base must be considered. Materials and methods There have been few reports in the Harmonious cranial growth is dependent on patent sutures, literature of isolated frontosphenoidal suture fusion, and and any craniosynostosis might lead to an asymmetrical we would like to report a series of five cases, as the shape of the skull. The anterior skull base is formed of recognition of this entity is important for its treatment. different bones, connected by sutures, fusing at different ages. The frontosphenoidal suture extends from the end of Presented at the Consensus Conference on Pediatric Neurosurgery, the frontoparietal suture, anteriorly and inferiorly in the Rome, 1–2 December 2006. -

Genetics of Congenital Hand Anomalies
G. C. Schwabe1 S. Mundlos2 Genetics of Congenital Hand Anomalies Die Genetik angeborener Handfehlbildungen Original Article Abstract Zusammenfassung Congenital limb malformations exhibit a wide spectrum of phe- Angeborene Handfehlbildungen sind durch ein breites Spektrum notypic manifestations and may occur as an isolated malforma- an phänotypischen Manifestationen gekennzeichnet. Sie treten tion and as part of a syndrome. They are individually rare, but als isolierte Malformation oder als Teil verschiedener Syndrome due to their overall frequency and severity they are of clinical auf. Die einzelnen Formen kongenitaler Handfehlbildungen sind relevance. In recent years, increasing knowledge of the molecu- selten, besitzen aber aufgrund ihrer Häufigkeit insgesamt und lar basis of embryonic development has significantly enhanced der hohen Belastung für Betroffene erhebliche klinische Rele- our understanding of congenital limb malformations. In addi- vanz. Die fortschreitende Erkenntnis über die molekularen Me- tion, genetic studies have revealed the molecular basis of an in- chanismen der Embryonalentwicklung haben in den letzten Jah- creasing number of conditions with primary or secondary limb ren wesentlich dazu beigetragen, die genetischen Ursachen kon- involvement. The molecular findings have led to a regrouping of genitaler Malformationen besser zu verstehen. Der hohe Grad an malformations in genetic terms. However, the establishment of phänotypischer Variabilität kongenitaler Handfehlbildungen er- precise genotype-phenotype correlations for limb malforma- schwert jedoch eine Etablierung präziser Genotyp-Phänotyp- tions is difficult due to the high degree of phenotypic variability. Korrelationen. In diesem Übersichtsartikel präsentieren wir das We present an overview of congenital limb malformations based Spektrum kongenitaler Malformationen, basierend auf einer ent- 85 on an anatomic and genetic concept reflecting recent molecular wicklungsbiologischen, anatomischen und genetischen Klassifi- and developmental insights. -

Osteodystrophy-Like Syndrome Localized to 2Q37
Am. J. Hum. Genet. 56:400-407, 1995 Brachydactyly and Mental Retardation: An Albright Hereditary Osteodystrophy-like Syndrome Localized to 2q37 L. C. Wilson,"7 K. Leverton,3 M. E. M. Oude Luttikhuis,' C. A. Oley,4 J. Flint,5 J. Wolstenholme,4 D. P. Duckett,2 M. A. Barrow,2 J. V. Leonard,6 A. P. Read,3 and R. C. Trembath' 'Departments of Genetics and Medicine, University of Leicester, and 2Leicestershire Genetics Centre, Leicester Royal Infirmary, Leicester, 3Department of Medical Genetics, St Mary's Hospital, Manchester. 4Department of Human Genetics, University of Newcastle upon Tyne, Newcastle upon Tyne; 5MRC Molecular Haematology Unit, Institute of Molecular Medicine, Oxford; and 6Medical Unit and 7Mothercare Unit for Clinical Genetics and Fetal Medicine, Institute of Child Health, London Summary The physical feature brachymetaphalangia refers to shortening of either the metacarpals and phalanges of the We report five patients with a combination ofbrachymeta- hands or of the equivalent bones in the feet. The combina- phalangia and mental retardation, similar to that observed tion of brachymetaphalangia and mental retardation occurs in Albright hereditary osteodystrophy (AHO). Four pa- in Albright hereditary osteodystrophy (AHO) (Albright et tients had cytogenetically visible de novo deletions of chro- al. 1942), a dysmorphic syndrome that is also associated mosome 2q37. The fifth patient was cytogenetically nor- with cutaneous ossification (in si60% of cases [reviewed by mal and had normal bioactivity of the a subunit of Gs Fitch 1982]); round face; and short, stocky build. Affected (Gsa), the protein that is defective in AHO. In this patient, individuals with AHO may have either pseudohypopara- we have used a combination of highly polymorphic molec- thyroidism (PHP), with end organ resistance to parathyroid ular markers and FISH to demonstrate a microdeletion at hormone (PTH) and certain other cAMP-dependent hor- 2q37. -

Orphanet Journal of Rare Diseases Biomed Central
Orphanet Journal of Rare Diseases BioMed Central Review Open Access Brachydactyly Samia A Temtamy* and Mona S Aglan Address: Department of Clinical Genetics, Human Genetics and Genome Research Division, National Research Centre (NRC), El-Buhouth St., Dokki, 12311, Cairo, Egypt Email: Samia A Temtamy* - [email protected]; Mona S Aglan - [email protected] * Corresponding author Published: 13 June 2008 Received: 4 April 2008 Accepted: 13 June 2008 Orphanet Journal of Rare Diseases 2008, 3:15 doi:10.1186/1750-1172-3-15 This article is available from: http://www.ojrd.com/content/3/1/15 © 2008 Temtamy and Aglan; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Brachydactyly ("short digits") is a general term that refers to disproportionately short fingers and toes, and forms part of the group of limb malformations characterized by bone dysostosis. The various types of isolated brachydactyly are rare, except for types A3 and D. Brachydactyly can occur either as an isolated malformation or as a part of a complex malformation syndrome. To date, many different forms of brachydactyly have been identified. Some forms also result in short stature. In isolated brachydactyly, subtle changes elsewhere may be present. Brachydactyly may also be accompanied by other hand malformations, such as syndactyly, polydactyly, reduction defects, or symphalangism. For the majority of isolated brachydactylies and some syndromic forms of brachydactyly, the causative gene defect has been identified. -

Appendix A: Organisation of a Craniofacial Unit
Appendix A: Organisation of a Craniofacial Unit The requirements of patients with craniofacial abnormalities are very complex and demand a multidisciplinary approach. Many body systems are affected, and every detail of patient management has to be given due attention. Care begins at birth and continues until the patient and his family have been relieved of the burden of the anomaly. A team is needed, capable of delivering expert patient care, and representative of all the relevant disciplines. Data, in the form of histories, physical examinations, and special investigations, are needed in planning treatment, and such data should be used to the maximum scientific effect, to improve present methods of management, still far from satisfactory, and to expand knowledge of the biology of cranial growth and its disorders. Craniofacial Units Sporadic craniofacial procedures performed by a surgeon on an irregular basis invite disaster. Tessier (1971a) estimated that each craniofacial centre should serve a population of 10 to 20 million people, provided that the team performed only craniofacial surgery and treated at least 50 new cases annually. As a consequence of Tessier's example and teaching there are now centres of acknowledged excellence in Paris and Nancy, attracting patients not only from France but also from North Africa and elsewhere. In North America there are now important craniofacial centres in Philadelphia, New York, Boston, Toronto, and Mexico City. Munro (1975) proposed that North America should be divided into seven regions, six for the United States and one for Canada, each serving populations of 20 to 40 million people. He believed that such centres would allow a concentration of multidisciplinary skills and accumulation of experience in the treatment of craniofacial anomalies. -

A Narrative Review of Poland's Syndrome
Review Article A narrative review of Poland’s syndrome: theories of its genesis, evolution and its diagnosis and treatment Eman Awadh Abduladheem Hashim1,2^, Bin Huey Quek1,3,4^, Suresh Chandran1,3,4,5^ 1Department of Neonatology, KK Women’s and Children’s Hospital, Singapore, Singapore; 2Department of Neonatology, Salmanya Medical Complex, Manama, Kingdom of Bahrain; 3Department of Neonatology, Duke-NUS Medical School, Singapore, Singapore; 4Department of Neonatology, NUS Yong Loo Lin School of Medicine, Singapore, Singapore; 5Department of Neonatology, NTU Lee Kong Chian School of Medicine, Singapore, Singapore Contributions: (I) Conception and design: EAA Hashim, S Chandran; (II) Administrative support: S Chandran, BH Quek; (III) Provision of study materials: EAA Hashim, S Chandran; (IV) Collection and assembly: All authors; (V) Data analysis and interpretation: BH Quek, S Chandran; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: A/Prof. Suresh Chandran. Senior Consultant, Department of Neonatology, KK Women’s and Children’s Hospital, Singapore 229899, Singapore. Email: [email protected]. Abstract: Poland’s syndrome (PS) is a rare musculoskeletal congenital anomaly with a wide spectrum of presentations. It is typically characterized by hypoplasia or aplasia of pectoral muscles, mammary hypoplasia and variably associated ipsilateral limb anomalies. Limb defects can vary in severity, ranging from syndactyly to phocomelia. Most cases are sporadic but familial cases with intrafamilial variability have been reported. Several theories have been proposed regarding the genesis of PS. Vascular disruption theory, “the subclavian artery supply disruption sequence” (SASDS) remains the most accepted pathogenic mechanism. Clinical presentations can vary in severity from syndactyly to phocomelia in the limbs and in the thorax, rib defects to severe chest wall anomalies with impaired lung function. -

TP63-Mutation As a Cause of Prenatal Lethal Multicystic Dysplastic Kidneys
Western University Scholarship@Western Paediatrics Publications Paediatrics Department 11-1-2020 TP63-mutation as a cause of prenatal lethal multicystic dysplastic kidneys Isabel Friedmann Carla Campagnolo Nancy Chan Ghislain Hardy Maha Saleh Follow this and additional works at: https://ir.lib.uwo.ca/paedpub Part of the Pediatrics Commons Received: 28 April 2020 | Revised: 8 August 2020 | Accepted: 10 August 2020 DOI: 10.1002/mgg3.1486 CLINICAL REPORT TP63-mutation as a cause of prenatal lethal multicystic dysplastic kidneys Isabel Friedmann1 | Carla Campagnolo2 | Nancy Chan1,3 | Ghislain Hardy1,4 | Maha Saleh1,2 1Schulich School of Medicine and Dentistry, University of Western Ontario, Abstract London, ON, Canada Background: Ectrodactyly-ectodermal dysplasia-clefting syndrome 3 (EEC) is 2Division of Genetics and Metabolism, one of the six overlapping syndromes caused by mutations in the tumor protein p63 Department of Paediatrics, London Health gene (TP63). EEC is suspected when patients have cleft hands or feet, polydactyly, Sciences Centre, London, ON, Canada 3 and syndactyly, abnormal development of the ectodermally derived structures, and Department of Pathology, London Health Sciences Centre, London, ON, Canada orofacial clefting. Genitourinary (GU) anomalies have been identified in patients 4Department of Obstetrics and Gynecology, with EEC, yet these are often under-recognized and under-reported. The available London Health Sciences Centre, London, literature on sonographic prenatal findings is sparse, especially when considering ON, Canada GU anomalies. Correspondence Methods: We present the case of a male stillborn fetus, who was found antenatally to Isabel Friedmann, University of Western have multicystic dysplastic kidneys and anhydramnios. Following the termination of Ontario, Schulich School of Medicine and Dentistry, London, Ontario, Canada. -

Classific-Ation and Identification of Inherited Brachydactylies
Journal of Medical Genetics, 1979, 16, 36-44 Classific-ation and identification of inherited brachydactylies NAOMI FITCH From the Lady Davis Institute for Medical Research, Jewish General Hospital, Montreal, Quebec, Canada SUMMARY A search for patterns of malformation in the brachydactylies has resulted in new ways to identify the different types. Type A-I can be characterised by a proportionate reduction of the middle phalanges. Type B is thought to be an amputation-like defect. In type C the fourth middle phalanx is usually the longest, and type E (Riccardi and Holmes, 1974) is characterised by short metacarpals and short distal phalanges. Short stature is usually present in type A-1 and type E brachydactyly (Riccardi and Holmes, 1974) and it may be present in some individuals with brachydactyly C. As short children have short hands, it is possible that in patients with very mild expressions of brachydactyly the cause of the short stature may be overlooked. It is suggested that in every child with propor- tionate short stature the hands should be carefully examined. If the hands are disproportionately short, if any distal creases are missing, if there is a shortening, however mild, of any finger, if any metacarpals are short, then it is important to have x-rays to look for brachydactyly A-1, C, or E. Much information is still needed. It is important in future reports to have skeletal surveys, pattern profile analyses, and to note the height of children with brachydactyly C. Most interesting of all will be when fetal limbs of each type become available for study. -

Free PDF Download
Eur opean Rev iew for Med ical and Pharmacol ogical Sci ences 2015; 19: 4549-4552 Concomitance of types D and E brachydactyly: a case report T. TÜLAY KOCA 1, F. ÇILEDA ğ ÖZDEMIR 2 1Malatya State Hospital, Physical Medicine and Rehabilitation Clinic, Malatya, Turkey 2Inonu University School of Medicine, Department of Physical Medicine and Rehabilitation, Malatya, Turkey Abstract. – Here, we present of a 35-year old examination, it was determined that the patient, female diagnosed with an overlapping form of who had kyphotic posture and brachydactyly in non-syndromic brachydactyly types D and E the 3 rd and 4 th finger of the right hand, in the 4th with phenotypic and radiological signs. There finger of the left hand and clinodactyly with was observed to be shortening in the right hand th metacarpal of 3 rd and 4 th fingers and left hand brachdactyly in the 4 toe of the left foot (Fig - metacarpal of 4 th finger and left foot metatarsal ures 1 and 2). It was learned that these deformi - of 4 th toe. There was also shortening of the distal ties had been present since birth and a younger phalanx of the thumbs and thoracic kyphosis. sister had similar shortness of the fingers. There The syndromic form of brachydactyly type E is was no known systemic disease. The menstrual firmly associated with pseudo-hypopthyroidism cycle was regular and there was no known his - as resistance to pthyroid hormone is the most prominent feature. As the patient had normal tory of osteoporosis. In the laboratory tests, the stature, normal laboratory parameters and no results of full blood count, sedimentation, psychomotor developmental delay, the case was parathormon (PTH), vitamin D, calcium, alka - classified as isolated E type brachydactyly. -

Holoprosencephaly and Preaxial Polydactyly Associated with a 1.24 Mb Duplication Encompassing FBXW11 at 5Q35.1
J Hum Genet (2006) 51:721–726 DOI 10.1007/s10038-006-0010-8 SHORT COMMUNICATION Holoprosencephaly and preaxial polydactyly associated with a 1.24 Mb duplication encompassing FBXW11 at 5q35.1 David A. Koolen Æ Jos Herbergs Æ Joris A. Veltman Æ Rolph Pfundt Æ Hans van Bokhoven Æ Hans Stroink Æ Erik A. Sistermans Æ Han G. Brunner Æ Ad Geurts van Kessel Æ Bert B. A. de Vries Received: 22 March 2006 / Accepted: 2 May 2006 / Published online: 25 July 2006 Ó The Japan Society of Human Genetics and Springer-Verlag 2006 Abstract Holoprosencephaly (HPE) is the most gion encompasses seven genes: RANBP17, TLX3, common developmental defect affecting the forebrain NPM1, FGF18, FBXW11, STK10, and DC-UbP. Since and midface in humans. The aetiology of HPE is highly FBXW11 is relatively highly expressed in fetal brain heterogeneous and includes both environmental and and is directly involved in proteolytic processing of genetic factors. Here we report on a boy with mild GLI3, we propose FBXW11 as the most likely candi- mental retardation, lobar HPE, epilepsy, mild pyra- date gene for the HPE and prexial polydactyly phe- midal syndrome of the legs, ventricular septal defect, notype. Additional research is needed to further vesicoureteral reflux, preaxial polydactyly, and facial establish the role of genes from the 5q35.1 region in dysmorphisms. Genome-wide tiling path resolution brain and limb development and to determine the array based comparative genomic hybridisation (array prevalence of copy number gain in the 5q35.1 region CGH) revealed a de novo copy-number gain at 5q35.1 among HPE patients. -

Prenatal Ultrasonography of Craniofacial Abnormalities
Prenatal ultrasonography of craniofacial abnormalities Annisa Shui Lam Mak, Kwok Yin Leung Department of Obstetrics and Gynaecology, Queen Elizabeth Hospital, Hong Kong SAR, China REVIEW ARTICLE https://doi.org/10.14366/usg.18031 pISSN: 2288-5919 • eISSN: 2288-5943 Ultrasonography 2019;38:13-24 Craniofacial abnormalities are common. It is important to examine the fetal face and skull during prenatal ultrasound examinations because abnormalities of these structures may indicate the presence of other, more subtle anomalies, syndromes, chromosomal abnormalities, or even rarer conditions, such as infections or metabolic disorders. The prenatal diagnosis of craniofacial abnormalities remains difficult, especially in the first trimester. A systematic approach to the fetal Received: May 29, 2018 skull and face can increase the detection rate. When an abnormality is found, it is important Revised: June 30, 2018 to perform a detailed scan to determine its severity and search for additional abnormalities. Accepted: July 3, 2018 Correspondence to: The use of 3-/4-dimensional ultrasound may be useful in the assessment of cleft palate and Kwok Yin Leung, MBBS, MD, FRCOG, craniosynostosis. Fetal magnetic resonance imaging can facilitate the evaluation of the palate, Cert HKCOG (MFM), Department of micrognathia, cranial sutures, brain, and other fetal structures. Invasive prenatal diagnostic Obstetrics and Gynaecology, Queen Elizabeth Hospital, Gascoigne Road, techniques are indicated to exclude chromosomal abnormalities. Molecular analysis for some Kowloon, Hong Kong SAR, China syndromes is feasible if the family history is suggestive. Tel. +852-3506 6398 Fax. +852-2384 5834 E-mail: [email protected] Keywords: Craniofacial; Prenatal; Ultrasound; Three-dimensional ultrasonography; Fetal structural abnormalities This is an Open Access article distributed under the Introduction terms of the Creative Commons Attribution Non- Commercial License (http://creativecommons.org/ licenses/by-nc/3.0/) which permits unrestricted non- Craniofacial abnormalities are common. -

An Isolated Case of Brachyphalangism of the Basal Finger Bones of the Little Finger with Symptoms of Tenosynovitis: a Case Report
Open Journal of Rheumatology and Autoimmune Diseases, 2018, 8, 66-70 http://www.scirp.org/journal/ojra ISSN Online: 2164-005X ISSN Print: 2163-9914 An Isolated Case of Brachyphalangism of the Basal Finger Bones of the Little Finger with Symptoms of Tenosynovitis: A Case Report Kazuhiko Hashimoto*, Ryosuke Kakinoki, Yukiko Hara, Naohiro Oka, Hiroki Tanaka, Kazuhiro Ohtani, Masao Akagi Department of Orthopedic Surgery, Kindai University Hospital, Osakasayama City, Osaka, Japan How to cite this paper: Hashimoto, K., Abstract Kakinoki, R., Hara, Y., Oka, N., Tanaka, H., Ohtani, K. and Akagi, M. (2018) An Isolated Brachydactyly is a general term that refers to disproportionately short fingers Case of Brachyphalangism of the Basal Fin- and toes and forms a part of the group of limb malformations characterized ger Bones of the Little Finger with Symp- by bone dysostosis. Brachydactyly usually occurs either as an isolated malfor- toms of Tenosynovitis: A Case Report. Open Journal of Rheumatology and Autoimmune mation or as a part of a complex malformation syndrome. Brachydactyly is Diseases, 8, 66-70. classified as types A, B, C, D, or E; brachymetatarsus IV; Sugarman brachy- https://doi.org/10.4236/ojra.2018.82006 dactyly; or Kirner deformity. Various types of isolated brachydactyly are rare, except for types A3 and D. We describe a 15-year-old girl with isolated bra- Received: April 7, 2018 Accepted: May 22, 2018 chyphalangism of the basal finger bones of the little finger with symptoms of Published: May 25, 2018 tenosynovitis. Tenosynovitis might be caused by growth deviation between the flexor digitorum superficialis and the flexor digitorum profundus.