UNIVERSITY of CALIFORNIA, SAN DIEGO Genome Assembly And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Grammar String: a Novel Ncrna Secondary Structure Representation
Grammar string: a novel ncRNA secondary structure representation Rujira Achawanantakun, Seyedeh Shohreh Takyar, and Yanni Sun∗ Department of Computer Science and Engineering, Michigan State University, East Lansing, MI 48824 , USA ∗Email: [email protected] Multiple ncRNA alignment has important applications in homologous ncRNA consensus structure derivation, novel ncRNA identification, and known ncRNA classification. As many ncRNAs’ functions are determined by both their sequences and secondary structures, accurate ncRNA alignment algorithms must maximize both sequence and struc- tural similarity simultaneously, incurring high computational cost. Faster secondary structure modeling and alignment methods using trees, graphs, probability matrices have thus been developed. Despite promising results from existing ncRNA alignment tools, there is a need for more efficient and accurate ncRNA secondary structure modeling and alignment methods. In this work, we introduce grammar string, a novel ncRNA secondary structure representation that encodes an ncRNA’s sequence and secondary structure in the parameter space of a context-free grammar (CFG). Being a string defined on a special alphabet constructed from a CFG, it converts ncRNA alignment into sequence alignment with O(n2) complexity. We align hundreds of ncRNA families from BraliBase 2.1 using grammar strings and compare their consensus structure with Murlet using the structures extracted from Rfam as reference. Our experiments have shown that grammar string based multiple sequence alignment competes favorably in consensus structure quality with Murlet. Source codes and experimental data are available at http://www.cse.msu.edu/~yannisun/grammar-string. 1. INTRODUCTION both the sequence and structural conservations. A successful application of SCFG is ncRNA classifica- Annotating noncoding RNAs (ncRNAs), which are tion, which classifies query sequences into annotated not translated into protein but function directly as ncRNA families such as tRNA, rRNA, riboswitch RNA, is highly important to modern biology. -

The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-Rnns and the Protein Structure Prediction Problem
Journal of Machine Learning Research 4 (2003) 575-602 Submitted 2/02; Revised 4/03; Published 9/03 The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-RNNs and the Protein Structure Prediction Problem Pierre Baldi [email protected] Gianluca Pollastri [email protected] School of Information and Computer Science Institute for Genomics and Bioinformatics University of California, Irvine Irvine, CA 92697-3425, USA Editor: Michael I. Jordan Abstract We describe a general methodology for the design of large-scale recursive neural network architec- tures (DAG-RNNs) which comprises three fundamental steps: (1) representation of a given domain using suitable directed acyclic graphs (DAGs) to connect visible and hidden node variables; (2) parameterization of the relationship between each variable and its parent variables by feedforward neural networks; and (3) application of weight-sharing within appropriate subsets of DAG connec- tions to capture stationarity and control model complexity. Here we use these principles to derive several specific classes of DAG-RNN architectures based on lattices, trees, and other structured graphs. These architectures can process a wide range of data structures with variable sizes and dimensions. While the overall resulting models remain probabilistic, the internal deterministic dy- namics allows efficient propagation of information, as well as training by gradient descent, in order to tackle large-scale problems. These methods are used here to derive state-of-the-art predictors for protein structural features such as secondary structure (1D) and both fine- and coarse-grained contact maps (2D). Extensions, relationships to graphical models, and implications for the design of neural architectures are briefly discussed. -

120421-24Recombschedule FINAL.Xlsx
Friday 20 April 18:00 20:00 REGISTRATION OPENS in Fira Palace 20:00 21:30 WELCOME RECEPTION in CaixaForum (access map) Saturday 21 April 8:00 8:50 REGISTRATION 8:50 9:00 Opening Remarks (Roderic GUIGÓ and Benny CHOR) Session 1. Chair: Roderic GUIGÓ (CRG, Barcelona ES) 9:00 10:00 Richard DURBIN The Wellcome Trust Sanger Institute, Hinxton UK "Computational analysis of population genome sequencing data" 10:00 10:20 44 Yaw-Ling Lin, Charles Ward and Steven Skiena Synthetic Sequence Design for Signal Location Search 10:20 10:40 62 Kai Song, Jie Ren, Zhiyuan Zhai, Xuemei Liu, Minghua Deng and Fengzhu Sun Alignment-Free Sequence Comparison Based on Next Generation Sequencing Reads 10:40 11:00 178 Yang Li, Hong-Mei Li, Paul Burns, Mark Borodovsky, Gene Robinson and Jian Ma TrueSight: Self-training Algorithm for Splice Junction Detection using RNA-seq 11:00 11:30 coffee break Session 2. Chair: Bonnie BERGER (MIT, Cambrige US) 11:30 11:50 139 Son Pham, Dmitry Antipov, Alexander Sirotkin, Glenn Tesler, Pavel Pevzner and Max Alekseyev PATH-SETS: A Novel Approach for Comprehensive Utilization of Mate-Pairs in Genome Assembly 11:50 12:10 171 Yan Huang, Yin Hu and Jinze Liu A Robust Method for Transcript Quantification with RNA-seq Data 12:10 12:30 120 Zhanyong Wang, Farhad Hormozdiari, Wen-Yun Yang, Eran Halperin and Eleazar Eskin CNVeM: Copy Number Variation detection Using Uncertainty of Read Mapping 12:30 12:50 205 Dmitri Pervouchine Evidence for widespread association of mammalian splicing and conserved long range RNA structures 12:50 13:10 169 Melissa Gymrek, David Golan, Saharon Rosset and Yaniv Erlich lobSTR: A Novel Pipeline for Short Tandem Repeats Profiling in Personal Genomes 13:10 13:30 217 Rory Stark Differential oestrogen receptor binding is associated with clinical outcome in breast cancer 13:30 15:00 lunch break Session 3. -

Methodology for Predicting Semantic Annotations of Protein Sequences by Feature Extraction Derived of Statistical Contact Potentials and Continuous Wavelet Transform
Universidad Nacional de Colombia Sede Manizales Master’s Thesis Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform Author: Supervisor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez A thesis submitted in fulfillment of the requirements for the degree of Master’s on Engineering - Industrial Automation in the Department of Electronic, Electric Engineering and Computation Signal Processing and Recognition Group June 2014 Universidad Nacional de Colombia Sede Manizales Tesis de Maestr´ıa Metodolog´ıapara predecir la anotaci´on sem´antica de prote´ınaspor medio de extracci´on de caracter´ısticas derivadas de potenciales de contacto y transformada wavelet continua Autor: Tutor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez Tesis presentada en cumplimiento a los requerimientos necesarios para obtener el grado de Maestr´ıaen Ingenier´ıaen Automatizaci´onIndustrial en el Departamento de Ingenier´ıaEl´ectrica,Electr´onicay Computaci´on Grupo de Procesamiento Digital de Senales Enero 2014 UNIVERSIDAD NACIONAL DE COLOMBIA Abstract Faculty of Engineering and Architecture Department of Electronic, Electric Engineering and Computation Master’s on Engineering - Industrial Automation Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform by Gustavo Alonso Arango Argoty In this thesis, a method to predict semantic annotations of the proteins from its primary structure is proposed. The main contribution of this thesis lies in the implementation of a novel protein feature representation, which makes use of the pairwise statistical contact potentials describing the protein interactions and geometry at the atomic level. -

Deep Learning in Chemoinformatics Using Tensor Flow
UC Irvine UC Irvine Electronic Theses and Dissertations Title Deep Learning in Chemoinformatics using Tensor Flow Permalink https://escholarship.org/uc/item/963505w5 Author Jain, Akshay Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, IRVINE Deep Learning in Chemoinformatics using Tensor Flow THESIS submitted in partial satisfaction of the requirements for the degree of MASTER OF SCIENCE in Computer Science by Akshay Jain Thesis Committee: Professor Pierre Baldi, Chair Professor Cristina Videira Lopes Professor Eric Mjolsness 2017 c 2017 Akshay Jain DEDICATION To my family and friends. ii TABLE OF CONTENTS Page LIST OF FIGURES v LIST OF TABLES vi ACKNOWLEDGMENTS vii ABSTRACT OF THE THESIS viii 1 Introduction 1 1.1 QSAR Prediction Methods . .2 1.2 Deep Learning . .4 2 Artificial Neural Networks(ANN) 5 2.1 Artificial Neuron . .5 2.2 Activation Function . .7 2.3 Loss function . .8 2.4 Optimization . .8 3 Deep Recursive Architectures 10 3.1 Recurrent Neural Networks (RNN) . 10 3.2 Recursive Neural Networks . 11 3.3 Directed Acyclic Graph Recursive Neural Networks (DAG-RNN) . 11 4 UG-RNN for small molecules 14 4.1 DAG Generation . 16 4.2 Local Information Vector . 16 4.3 Contextual Vectors . 17 4.4 Activity Prediction . 17 4.5 UG-RNN With Contracted Rings (UG-RNN-CR) . 18 4.6 Example: UG-RNN Model of Propionic Acid . 20 5 Implementation 24 6 Data & Results 26 6.1 Aqueous Solubility Prediction . 26 6.2 Melting Point Prediction . 28 iii 7 Conclusions 30 Bibliography 32 A Source Code 37 A.1 UGRNN . -

Course Outline
Department of Computer Science and Software Engineering COMP 6811 Bioinformatics Algorithms (Reading Course) Fall 2019 Section AA Instructor: Gregory Butler Curriculum Description COMP 6811 Bioinformatics Algorithms (4 credits) The principal objectives of the course are to cover the major algorithms used in bioinformatics; sequence alignment, multiple sequence alignment, phylogeny; classi- fying patterns in sequences; secondary structure prediction; 3D structure prediction; analysis of gene expression data. This includes dynamic programming, machine learning, simulated annealing, and clustering algorithms. Algorithmic principles will be emphasized. A project is required. Outline of Topics The course will focus on algorithms for protein sequence analysis. It will not cover genome assembly, genome mapping, or gene recognition. • Background in Biology and Genomics • Sequence Alignment: Pairwise and Multiple • Representation of Protein Amino Acid Composition • Profile Hidden Markov Models • Specificity Determining Sites • Curation, Annotation, and Ontologies • Machine Learning: Secondary Structure, Signals, Subcellular Location • Protein Families, Phylogenomics, and Orthologous Groups • Profile-Based Alignments • Algorithms Based on k-mers 1 Texts | in Library D. Higgins and W. Taylor (editors). Bioinformatics: Sequence, Structure and Databanks, Oxford University Press, 2000. A. D. Baxevanis and B. F. F. Ouelette. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins, Wiley, 1998. Richard Durbin, Sean R. Eddy, Anders Krogh, -

Deep Learning in Science Pierre Baldi Frontmatter More Information
Cambridge University Press 978-1-108-84535-9 — Deep Learning in Science Pierre Baldi Frontmatter More Information Deep Learning in Science This is the first rigorous, self-contained treatment of the theory of deep learning. Starting with the foundations of the theory and building it up, this is essential reading for any scientists, instructors, and students interested in artificial intelligence and deep learning. It provides guidance on how to think about scientific questions, and leads readers through the history of the field and its fundamental connections to neuro- science. The author discusses many applications to beautiful problems in the natural sciences, in physics, chemistry, and biomedicine. Examples include the search for exotic particles and dark matter in experimental physics, the prediction of molecular properties and reaction outcomes in chemistry, and the prediction of protein structures and the diagnostic analysis of biomedical images in the natural sciences. The text is accompanied by a full set of exercises at different difficulty levels and encourages out-of-the-box thinking. Pierre Baldi is Distinguished Professor of Computer Science at University of California, Irvine. His main research interest is understanding intelligence in brains and machines. He has made seminal contributions to the theory of deep learning and its applications to the natural sciences, and has written four other books. © in this web service Cambridge University Press www.cambridge.org Cambridge University Press 978-1-108-84535-9 — Deep Learning in -

Michael S. Waterman: Breathing Mathematics Into Genes >>>
ISSUE 13 Newsletter of Institute for Mathematical Sciences, NUS 2008 Michael S. Waterman: Breathing Mathematics into Genes >>> setting up of the Center for Computational and Experimental Genomics in 2001, Waterman and his collaborators and students continue to provide a road map for the solution of post-genomic computational problems. For his scientific contributions he was elected fellow or member of prestigious learned bodies like the American Academy of Arts and Sciences, National Academy of Sciences, American Association for the Advancement of Science, Institute of Mathematical Statistics, Celera Genomics and French Acadèmie des Sciences. He was awarded a Gairdner Foundation International Award and the Senior Scientist Accomplishment Award of the International Society of Computational Biology. He currently holds an Endowed Chair at USC and has held numerous visiting positions in major universities. In addition to research, he is actively involved in the academic and social activities of students as faculty master Michael Waterman of USC’s International Residential College at Parkside. Interview of Michael S. Waterman by Y.K. Leong Waterman has served as advisor to NUS on genomic research and was a member of the organizational committee Michael Waterman is world acclaimed for pioneering and of the Institute’s thematic program Post-Genome Knowledge 16 fundamental work in probability and algorithms that has Discovery (Jan – June 2002). On one of his advisory tremendous impact on molecular biology, genomics and visits to NUS, Imprints took the opportunity to interview bioinformatics. He was a founding member of the Santa him on 7 February 2007. The following is an edited and Cruz group that launched the Human Genome Project in enhanced version of the interview in which he describes the 1990, and his work was instrumental in bringing the public excitement of participating in one of the greatest modern and private efforts of mapping the human genome to their scientific adventures and of unlocking the mystery behind completion in 2003, two years ahead of schedule. -

Bringing Folding Pathways Into Strand Pairing Prediction
Bringing folding pathways into strand pairing prediction Jieun Jeong1,2, Piotr Berman1, and Teresa Przytycka2 1 Computer Science and Engineering Department The Pennsylvania State University University Park, PA 16802 USA 2 National Center for Biotechnology Information US National Library of Medicine, National Institutes of Health Bethesda, MD 20894 email: [email protected], [email protected], [email protected] Abstract. The topology of β-sheets is defined by the pattern of hydrogen- bonded strand pairing. Therefore, predicting hydrogen bonded strand partners is a fundamental step towards predicting β-sheet topology. In this work we report a new strand pairing algorithm. Our algorithm at- tempts to mimic elements of the folding process. Namely, in addition to ensuring that the predicted hydrogen bonded strand pairs satisfy basic global consistency constraints, it takes into account hypothetical folding pathways. Consistently with this view, introducing hydrogen bonds be- tween a pair of strands changes the probabilities of forming other strand pairs. We demonstrate that this approach provides an improvement over previously proposed algorithms. 1 Introduction The prediction of protein structure from protein sequence is a long-held goal that would provide invaluable information regarding the function of individ- ual proteins and the evolution of protein families. The increasing amount of sequence and structure data, made it possible to decouple the structure predic- tion problem from the problem of modeling of protein folding process. Indeed, a significant progress has been achieved by bioinformatics approaches such as homology modeling, threading, and assembly from fragments [16]. At the same time, the fundamental problem of how actually a protein acquires its final folded state remains a subject of controversy. -
ACDL-2020-Programme-Ver10.0
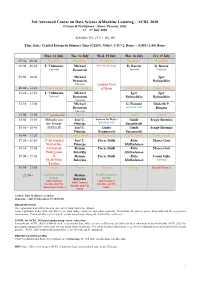
3rd Advanced Course on Data Science &Machine Learning – ACDL 2020 Certosa di Pontignano - Siena, Tuscany, Italy 13 – 17 July 2020 Schedule Ver. 11.0 – July 9th Time Zone: Central European Summer Time (CEST), Offset: UTC+2, Rome – (GMT+2:00) Rome Mon, 13 July Tue, 14 July Wed, 15 July Thu, 16 July Fri, 17 July 07:30 – 09:00 Breakfast Breakfast Breakfast Breakfast Breakfast 09:00 – 09:50 T. Viehmann Michael 8:30 Social Tour D. Bacciu D. Bacciu Tutorial Bronstein Tutorial Tutorial 09:50 – 10:40 Michael Igor Bronstein Babuschkin Tutorial Guided Visit 10:40 – 11:20 Coffee break Coffee break of Siena Coffee break Coffee break 11:20 – 12:10 T. Viehmann Michael Igor Igor Tutorial Bronstein Babuschkin Babuschkin Tutorial 12:10 – 13:00 Michael G. Fiameni Diederik P. Bronstein Industrial Talk Kingma Tutorial 13:00 – 15:00 Lunch (13:00-14:00) Lunch Lunch Lunch Lunch 15:00 – 15:50 Mihaela van José C. Lorenzo De Mattei Guido Sergiy Butenko der Schaar Principe Industrial Talk Sanguinetti 15:50 – 16:40 (14:00-16:40) José C. Guido Guido Sergiy Butenko Principe Sanguinetti Sanguinetti 16:40 – 17:20 Coffee break Coffee break Coffee break Coffee break Coffee break 17:20 – 18:10 17:20 Guided José C. Pierre Baldi Risto Marco Gori Visit of the Principe Miikkulainen 18:10 – 19:00 Certosa di Roman Pierre Baldi Risto Marco Gori Pontignano Belavkin Miikkulainen 19:00 – 19:50 & Roman Pierre Baldi Risto Varun Ojha 18:20 Wine Belavkin Miikkulainen Tutorial Tasting 19:50 – 21:50 Dinner Dinner Dinner Dinner Social Dinner 21:50 – Oral Presentation Roman Oral Presentation Session Belavkin Session with Cantucci with Cantucci with Cantucci biscuits and Vin biscuits and Vin biscuits and Vin Santo (sweet wine) Santo Santo Arrival: July 12 (Dinner at 20:30) Departure: July 18 (Breakfast 07:30-09:00) REGISTRATION The registration desk will be located close to the Main Conference Room. -

Gene and Genome Duplication David Sankoff
681 Gene and genome duplication David Sankoff Genomic sequencing projects have revealed the productivity of tetraploidization. I also summarize some mathematical processes duplicating genes or entire chromosome segments. modeling and algorithmics inspired by duplication phenomena. Substantial proportions of the yeast, Arabidopsis and human gene complements are made up of duplicates. This has prompted much Gene duplication interest in the processes of duplication, functional divergence and Li et al. [2] find that duplicated genes, as identified through loss of genes, has renewed the debate on whether an early fairly selective criteria, account for ~15% of the protein genes vertebrate genome was tetraploid, and has inspired mathematical in the human genome (counting both genes in each pair). In a models and algorithms in computational biology. survey of eukaryotic genome sequences, Lynch and Conery [3••], using a somewhat different filter, accounted for ~8%, Addresses 10% and 20% of the gene complement of the fly, yeast and Centre de recherches mathématiques, Université de Montréal, worm genomes, respectively. (Other estimates put the figure CP 6128 succursale Centre-Ville, Montreal, Québec H3C 3J7, Canada; at 16% for yeast and 25% for Arabidopsis [4•].) They estimated e-mail: [email protected] highly variable rates of gene duplication, averaging ~0.01 per Current Opinion in Genetics & Development 2001, 11:681–684 gene per Myr (million years). On the basis of ratios of silent and replacement rearrangements, they found that there is 0959-437X/01/$ — see front matter © 2001 Elsevier Science Ltd. All rights reserved. typically a period of neutral or (occasionally) even slightly accelerated evolution, lasting a few Myr at most, with one of Abbreviation the copies eventually being silenced in a large majority of Myr million years cases, and the remaining ones undergoing relatively stringent purifying selection. -

I S C B N E W S L E T T
ISCB NEWSLETTER FOCUS ISSUE {contents} President’s Letter 2 Member Involvement Encouraged Register for ISMB 2002 3 Registration and Tutorial Update Host ISMB 2004 or 2005 3 David Baker 4 2002 Overton Prize Recipient Overton Endowment 4 ISMB 2002 Committees 4 ISMB 2002 Opportunities 5 Sponsor and Exhibitor Benefits Best Paper Award by SGI 5 ISMB 2002 SIGs 6 New Program for 2002 ISMB Goes Down Under 7 Planning Underway for 2003 Hot Jobs! Top Companies! 8 ISMB 2002 Job Fair ISCB Board Nominations 8 Bioinformatics Pioneers 9 ISMB 2002 Keynote Speakers Invited Editorial 10 Anna Tramontano: Bioinformatics in Europe Software Recommendations11 ISCB Software Statement volume 5. issue 2. summer 2002 Community Development 12 ISCB’s Regional Affiliates Program ISCB Staff Introduction 12 Fellowship Recipients 13 Awardees at RECOMB 2002 Events and Opportunities 14 Bioinformatics events world wide INTERNATIONAL SOCIETY FOR COMPUTATIONAL BIOLOGY A NOTE FROM ISCB PRESIDENT This newsletter is packed with information on development and dissemination of bioinfor- the ISMB2002 conference. With over 200 matics. Issues arise from recommendations paper submissions and over 500 poster submis- made by the Society’s committees, Board of sions, the conference promises to be a scientific Directors, and membership at large. Important feast. On behalf of the ISCB’s Directors, staff, issues are defined as motions and are discussed EXECUTIVE COMMITTEE and membership, I would like to thank the by the Board of Directors on a bi-monthly Philip E. Bourne, Ph.D., President organizing committee, local organizing com- teleconference. Motions that pass are enacted Michael Gribskov, Ph.D., mittee, and program committee for their hard by the Executive Committee which also serves Vice President work preparing for the conference.