The Whale Shark Genome Reveals How Genomic and Physiological Properties Scale with Body Size
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using Information in Taxonomists' Heads to Resolve Hagfish And
This article was downloaded by: [Max Planck Inst fuer Evolutionsbiologie] On: 03 September 2013, At: 07:01 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Historical Biology: An International Journal of Paleobiology Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/ghbi20 Using information in taxonomists’ heads to resolve hagfish and lamprey relationships and recapitulate craniate–vertebrate phylogenetic history Maria Abou Chakra a , Brian Keith Hall b & Johnny Ricky Stone a b c d a Department of Biology , McMaster University , Hamilton , Canada b Department of Biology , Dalhousie University , Halifax , Canada c Origins Institute, McMaster University , Hamilton , Canada d SHARCNet, McMaster University , Hamilton , Canada Published online: 02 Sep 2013. To cite this article: Historical Biology (2013): Using information in taxonomists’ heads to resolve hagfish and lamprey relationships and recapitulate craniate–vertebrate phylogenetic history, Historical Biology: An International Journal of Paleobiology To link to this article: http://dx.doi.org/10.1080/08912963.2013.825792 PLEASE SCROLL DOWN FOR ARTICLE Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. -

Elasmobranch Biodiversity, Conservation and Management Proceedings of the International Seminar and Workshop, Sabah, Malaysia, July 1997
The IUCN Species Survival Commission Elasmobranch Biodiversity, Conservation and Management Proceedings of the International Seminar and Workshop, Sabah, Malaysia, July 1997 Edited by Sarah L. Fowler, Tim M. Reed and Frances A. Dipper Occasional Paper of the IUCN Species Survival Commission No. 25 IUCN The World Conservation Union Donors to the SSC Conservation Communications Programme and Elasmobranch Biodiversity, Conservation and Management: Proceedings of the International Seminar and Workshop, Sabah, Malaysia, July 1997 The IUCN/Species Survival Commission is committed to communicate important species conservation information to natural resource managers, decision-makers and others whose actions affect the conservation of biodiversity. The SSC's Action Plans, Occasional Papers, newsletter Species and other publications are supported by a wide variety of generous donors including: The Sultanate of Oman established the Peter Scott IUCN/SSC Action Plan Fund in 1990. The Fund supports Action Plan development and implementation. To date, more than 80 grants have been made from the Fund to SSC Specialist Groups. The SSC is grateful to the Sultanate of Oman for its confidence in and support for species conservation worldwide. The Council of Agriculture (COA), Taiwan has awarded major grants to the SSC's Wildlife Trade Programme and Conservation Communications Programme. This support has enabled SSC to continue its valuable technical advisory service to the Parties to CITES as well as to the larger global conservation community. Among other responsibilities, the COA is in charge of matters concerning the designation and management of nature reserves, conservation of wildlife and their habitats, conservation of natural landscapes, coordination of law enforcement efforts as well as promotion of conservation education, research and international cooperation. -

300556969.Pdf
ARTICLE https://doi.org/10.1038/s42003-019-0713-y OPEN The vertebrate Aqp14 water channel is a neuropeptide-regulated polytransporter François Chauvigné1, Ozlem Yilmaz2, Alba Ferré1, Per Gunnar Fjelldal3, Roderick Nigel Finn 1,2*& Joan Cerdà 1* 1234567890():,; Water channels (aquaporins) were originally discovered in mammals with fourteen sub- families now identified (AQP0-13). Here we show that a functional Aqp14 subfamily phy- logenetically related to AQP4-type channels exists in all vertebrate lineages except hagfishes and eutherian mammals. In contrast to the water-selective classical aquaporins, which have four aromatic-arginine constriction residues, Aqp14 proteins present five non-aromatic constriction residues and facilitate the permeation of water, urea, ammonia, H2O2 and gly- cerol. Immunocytochemical assays suggest that Aqp14 channels play important osmor- egulatory roles in piscine seawater adaptation. Our data indicate that Aqp14 intracellular trafficking is tightly regulated by the vasotocinergic/isotocinergic neuropeptide and receptor systems, whereby protein kinase C and A transduction pathways phosphorylate highly conserved C-terminal residues to control channel plasma membrane insertion. The neuro- peptide regulation of Aqp14 channels thus predates the vasotocin/vasopressin regulation of AQP2-5-6 orthologs observed in tetrapods. These findings demonstrate that vertebrate Aqp14 channels represent an ancient subfamily of neuropeptide-regulated polytransporters. 1 IRTA-Institute of Biotechnology and Biomedicine (IBB), Universitat -

Abstracts Part 1
375 Poster Session I, Event Center – The Snowbird Center, Friday 26 July 2019 Maria Sabando1, Yannis Papastamatiou1, Guillaume Rieucau2, Darcy Bradley3, Jennifer Caselle3 1Florida International University, Miami, FL, USA, 2Louisiana Universities Marine Consortium, Chauvin, LA, USA, 3University of California, Santa Barbara, Santa Barbara, CA, USA Reef Shark Behavioral Interactions are Habitat Specific Dominance hierarchies and competitive behaviors have been studied in several species of animals that includes mammals, birds, amphibians, and fish. Competition and distribution model predictions vary based on dominance hierarchies, but most assume differences in dominance are constant across habitats. More recent evidence suggests dominance and competitive advantages may vary based on habitat. We quantified dominance interactions between two species of sharks Carcharhinus amblyrhynchos and Carcharhinus melanopterus, across two different habitats, fore reef and back reef, at a remote Pacific atoll. We used Baited Remote Underwater Video (BRUV) to observe dominance behaviors and quantified the number of aggressive interactions or bites to the BRUVs from either species, both separately and in the presence of one another. Blacktip reef sharks were the most abundant species in either habitat, and there was significant negative correlation between their relative abundance, bites on BRUVs, and the number of grey reef sharks. Although this trend was found in both habitats, the decline in blacktip abundance with grey reef shark presence was far more pronounced in fore reef habitats. We show that the presence of one shark species may limit the feeding opportunities of another, but the extent of this relationship is habitat specific. Future competition models should consider habitat-specific dominance or competitive interactions. -

The Biology of Hagfishes the Biology of Hagfishes
THE BIOLOGY OF HAGFISHES THE BIOLOGY OF HAGFISHES J0RGEN M0RUP J0RGENSEN, JENS PETER LOMHOLT, ROY E. WEBER and HANSMALTE Department of Zoophysiology Institute of Biological Sciences University of Aarhus Denmark luni Springer-Science+Business Media, B.V. Published by Chapman and HaU, an imprint of Thomson Science, 2--6 Boundary Row, London SEl 8HN, UK Thomson Science, 2-6 Boundary Row, London SEl 8HN, UK Thomson Science, 115 Fifth Avenue, New York, NY 10003, USA Thomson Science, Suite 750, 400 Market Street, Philadelphia, PA 19106, USA Thomson Science, Pappelallee 3, 694469 Weinheim, Germany First edition 1998 © 1998 Springer Science+ Business Media Dordrecht OriginaUy published by Chapman & HaU Ltdin 1998 Softcover reprint of the hardcover 1st edition 1998 Typeset in 10112pt Palatino by Cambrian Typesetters, Frimley, Surrey ISBN 978-94-010-6465-1 ISBN 978-94-011-5834-3 (eBook) DOI 10.1007/978-94-011-5834-3 AII rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, e1e~tronic, mechanical, photocopying, recording or otherwise, without the prior written permis sion of the publishers. Applications for permission shou1d be addressed to the rights manager at the London address of the publisher. The publisher makes no representation, express or imp1ied, with regard to the accuracy of the information contained in this book and cannot accept any legal responsibility or liability for any errors or omissions that may be made. A catalogue record for this book is available from the British Library Library ofCongress Catalog Card Number: 97-69524 8 Printed on acid-free text paper, manufactured in accordance with ANSIINISO Z39.48-1992 (Permanence of Paper). -

Cartilaginous Fishes Offer Unique Insights Into the Evolution of The
General and Comparative Endocrinology 295 (2020) 113527 Contents lists available at ScienceDirect General and Comparative Endocrinology journal homepage: www.elsevier.com/locate/ygcen Research paper Cartilaginous fishes offer unique insights into the evolution of the nuclear receptor gene repertoire in gnathostomes T Elza Fonsecaa,b, André M. Machadoa, Nair Vilas-Arrondoc,d, André Gomes-dos-Santosa,b, Ana Veríssimob,e, Pedro Estevesb,d, Tereza Almeidab,e, Gonçalo Themudoa, Raquel Ruivoa, Montse Pérezc, Rute da Fonsecaf, Miguel M. Santosa,b, Elsa Froufea, Esther Román-Marcotec, ⁎ ⁎ Byrappa Venkateshg, , L. Filipe C. Castroa,b, a CIIMAR/CIMAR – Interdisciplinary Centre of Marine and Environmental Research, U.Porto, 4450-208 Matosinhos, Portugal b FCUP – Faculty of Sciences, Department of Biology, U.Porto, 4169-007 Porto, Portugal c AQUACOV, Instituto Español de Oceanografía, Centro Oceanográfico de Vigo, 36390 Vigo, Spain d UVIGO, phD Program “Marine Science, Tehchology and Management” (Do *MAR), Faculty of Biology, University of Vigo, 36200 Vigo, Spain e CIBIO – Research Center in Biodiversity and Genetic Resources, InBIO, Associate Laboratory, U.Porto, 4485-661 Vairão, Portugal f Center for Macroecology, Evolution and Climate, GLOBE Institute, University of Copenhagen, Denmark g Comparative Genomics Laboratory, Institute of Molecular and Cell Biology, A*STAR (Agency for Science, Technology and Research), Biopolis, Singapore 138673, Singapore ARTICLE INFO ABSTRACT Keywords: Nuclear receptors (NRs) are key transcription factors that originated in the common ancestor of metazoans. The Nuclear receptors vast majority of NRs are triggered by binding to either endogenous (e.g. retinoic acid) or exogenous (e.g. xe- Genome nobiotics) ligands, and their evolution and expansion is tightly linked to the function of endocrine systems. -
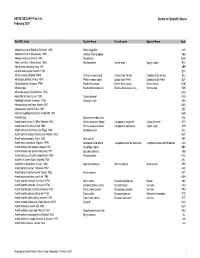
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

And Their Functional, Ecological, and Evolutionary Implications
DePaul University Via Sapientiae College of Science and Health Theses and Dissertations College of Science and Health Spring 6-14-2019 Body Forms in Sharks (Chondrichthyes: Elasmobranchii), and Their Functional, Ecological, and Evolutionary Implications Phillip C. Sternes DePaul University, [email protected] Follow this and additional works at: https://via.library.depaul.edu/csh_etd Part of the Biology Commons Recommended Citation Sternes, Phillip C., "Body Forms in Sharks (Chondrichthyes: Elasmobranchii), and Their Functional, Ecological, and Evolutionary Implications" (2019). College of Science and Health Theses and Dissertations. 327. https://via.library.depaul.edu/csh_etd/327 This Thesis is brought to you for free and open access by the College of Science and Health at Via Sapientiae. It has been accepted for inclusion in College of Science and Health Theses and Dissertations by an authorized administrator of Via Sapientiae. For more information, please contact [email protected]. Body Forms in Sharks (Chondrichthyes: Elasmobranchii), and Their Functional, Ecological, and Evolutionary Implications A Thesis Presented in Partial Fulfilment of the Requirements for the Degree of Master of Science June 2019 By Phillip C. Sternes Department of Biological Sciences College of Science and Health DePaul University Chicago, Illinois Table of Contents Table of Contents.............................................................................................................................ii List of Tables..................................................................................................................................iv -

Fishfaunistical Monitoring of the Hungarian Part of the River Drava (1999-2004)
Natura Somogyiensis 7 75-104 Kaposvár, 2005 Fishfaunistical monitoring of the Hungarian part of the River Drava (1999-2004) SALLAI ZOLTÁN &KONTOS TIVADAR NIMFEAEnvironmental and Nature Conservation Association, Szarvas H-5540, Hungary, P.O.Box: 122. SALLAI, Z. & KONTOS, T.: Fishfaunistical monitoring of the Hungarian part of the River Drava (1999-2004) Abstract: Fishfaunistical monitoring in the Hungarian part of the river Drava between 1999 and 2004 wa car- ried out. Asmall-capacity, pulsating direct current electric fishing machine with rechargeable battery was used for the surveys. During the monitoring altogether 22.649 fish specimen were caught and identified, represent- ing 44 species. Beyond the monitoring sites, fishfaunistical data were collected in other types of habitats, in side arms, the main channel and backwater arms as well. In addition, our own data was complemented with verified data supported by evidence species or picture documentation regarding the occurrence of species, and also with catching data from the fishery database, so the presence of altogether 57 species has been confirmed. Out of the 57 species of verified occurrence, 23 species have nature conservation status. 5 of these protected species have highest level protection status (Eudontomyzon mariae, Hucho hucho, Umbra krameri, Zingel zin- gel, Zingel streber). Out of the identified species 22 species are listed in the Annexes of the Habitat Directive. Based on the number of species found, the absolute (TA: 114) and relative natural value (TR: 2.036) of the fish fauna was defined. The recent fauna list of the river has been compiled using literature and own data; based on this, the occasional or regular occurrence of altogether 63 species is presumable in the river Drava. -

Agnatha, Chondrichthyes and Osteichthyes
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/311861377 Agnatha, Chondrichthyes and Osteichthyes Chapter · November 2016 CITATIONS READS 0 1,531 2 authors: Antonis Petrou Charitos Zapitis AP Marine Env.Consultancy Ltd & Enalia Physis Environmental Research Centre University of Derby 29 PUBLICATIONS 131 CITATIONS 1 PUBLICATION 0 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Lionfish in the Mediterranean View project PCY1 - Investigating the effects of recreational diving on the macroalgal communities of the 'Zenobia' shipwreck (Cyprus) View project All content following this page was uploaded by Charitos Zapitis on 24 December 2016. The user has requested enhancement of the downloaded file. Chapter 26 - Agnatha CHAPTER 26: AGNATHA, CHONDRICHTHYES AND OSTEICHTHYES FISHES Antonis Petrou and Charitos Zapitis 1. INTRODUCTION The classification of fishes is not straightforward since they do not form a natural scientific grouping like the other vertebrate classes, i.e. the amphibians, reptiles, birds and mammals. Indeed, fishes can be considered by exclusion to be vertebrates that are not tetrapods (see Chapter 25: Introduction to Vertebrates). They are aquatic, gill- bearing, ectothermic ('cold-blooded') animals with a distinguishable head and, when present, digit-less limbs. Traditionally, fish have been arranged into three groups: ➵ Agnatha 1, the jawless fish (Myxini [hagfishes] and Hyperoartia [lampreys]); ➵ Chondrichthyes, the cartilaginous fish (sharks, skates and rays); ➵ Osteichthyes, the bony fish (Actinopterygii [ray-finned fishes] and Sarcopterygii [lobe-finned fishes]). This classification is adequate for general purposes, although Agnatha is paraphyletic and includes several groups of extinct jawless fishes. -

A Lamprey from the Devonian Period of South Africa
Vol 000 | 00 Month 2006 | doi:10.1038/nature05150 LETTERS ; A lamprey from the Devonian period of South Africa < Robert W. Gess1, Michael I. Coates2 & Bruce S. Rubidge1 Lampreys are the most scientifically accessible of the remaining and the disc is proportionately larger than those of similarly sized, jawless vertebrates, but their evolutionary history is obscure. In post-metamorphic, living forms (Fig. 2). In contrast, the oral disc of contrast to the rich fossil record of armoured jawless fishes, all of the Late Carboniferous lamprey Mayomyzon7,8, if present, is much which date from the Devonian period and earlier1–3, only two smaller9, and no remnant of a disc is preserved in the Early Palaeozoic lampreys have been recorded, both from the Carboniferous lamprey Hardistiella10. The lamprey identity of the Carboniferous period1. In addition to these, the recent report of putative oral disc of Pipiscus11 is uncertain12. an exquisitely preserved Lower Cretaceous example4 demonstrates Priscomyzon displays a set of 14 evenly spaced teeth surrounding that anatomically modern lampreys were present by the late the mouth. These are the first teeth to be discovered in any fossil Mesozoic era. Here we report a marine/estuarine fossil lamprey lamprey, and resemble the circumoral arrangements of 19 or more from the Famennian (Late Devonian) of South Africa5,6, the iden- teeth present in modern forms such as Ichthyomyzon, Petromyzon, tity of which is established easily because many of the key specia- Caspiomyzon and Geotria13.InPriscomyzon, the posterior circumoral lizations of modern forms are already in place. These teeth appear to be more elongate than the remainder, whereas in specializations include the first evidence of a large oral disc, the modern forms lateral or anterior teeth tend to be largest. -

Independent Hox-Cluster Duplications in Lampreys
Independent Hox-Cluster Duplications in Lampreys Claudia Friedz;y, Sonja J. Prohaskaz;y, and Peter F. Stadlerz;y zBioinformatik, Institut fur¨ Informatik, Universit¨at Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany yInstitut fur¨ Theoretische Chemie und Molekulare Strukturbiologie Universit¨at Wien, W¨ahringerstraße 17, A-1090 Wien, Austria ∗Address for correspondence: Peter F. Stadler, Bioinformatik, Institut fur¨ Informatik, Universit¨at Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany. Phone: ++49 341 149 5120; Fax: ++49 341 149 5119; Email: [email protected]. Abstract. The analysis of the publicly available Hox gene sequences from the sea lamprey Petromyzon marinus provides evidence that the Hox clusters in lampreys and other vertebrate species arose from independent duplica- tions. In particular, our analysis supports the hypothesis that the last comman ancestor of agnathans and gnathostomes had only a single Hox cluster which was subsequently duplicated independently in the two lin- eages. Keywords. Hox clusters, lamprey, phylogenetic footprints 1 Fried et al.: Duplications of Lamprey Hox Clusters 2 1. Introduction Hox genes code for homeodomain containing transcription factors which are homolo- gous to the genes in the Drosophila homeotic gene clusters (McGinnis and Krumlauf, 1992; Schubert et al., 1993). There is good evidence that the common ancestor of sharks, bony fish, and tetrapods, had four clusters homologous to the mammalian ones (Holland and Garcia-Fernandez, 1996; Prohaska et al., 2003b). An additional duplication event in the teleost lineage increased the number of distinct clusters to at least 7, e.g. in zebrafish (Amores et al., 1998; Stellwag, 1999). The agnathan vertebrates, lampreys (Hyperoartia) and hagfishes (Hyperotreti), as the most primitive extant true vertebrates, occupy a phylogenetically intermediate position between the cephalochordates, such as amphioxus, with a single Hox clus- ter (Garcia-Fern´andez and Holland, 1994) and the gnathostomes with four or more clusters.