Investigation of Non-Local Autoionization Processes in Rare Gas Clusters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ettore Majorana and the Birth of Autoionization
Ettore Majorana and the birth of autoionization E. Arimondo∗,† Charles W. Clark,‡ and W. C. Martin§ National Institute of Standards and Technology Gaithersburg, MD 20899, USA (Dated: May 19, 2009) Abstract In some of the first applications of modern quantum mechanics to the spectroscopy of many-electron atoms, Ettore Majorana solved several outstanding problems by developing the theory of autoionization. Later literature makes only sporadic refer- ences to this accomplishment. After reviewing his work in its contemporary context, we describe subsequent developments in understanding the spectra treated by Ma- jorana, and extensions of his theory to other areas of physics. We find many puzzles concerning the way in which the modern theory of autoionization was developed. ∗ Permanent address: Dipartimento di Fisica E. Fermi, Universit`adi Pisa, Italy †Electronic address: [email protected] ‡Electronic address: [email protected] §Electronic address: [email protected] 1 Contents I.Introduction 2 II.TheState of Atomic Spectroscopy circa 1931 4 A.Observed Spectra 4 B.Theoriesof Unstable Electronic States 6 III.Symmetry Considerations for Doubly-Excited States 7 IV.Analyses of the Observed Double-Excitation Spectra 8 A.Double Excitation in Helium 8 B.TheIncomplete np2 3P Terms in Zinc, Cadmium, and Mercury 9 V.Contemporary and Subsequent Work on Autoionization 12 A.Shenstone’s Contemporary Identification of Autoionization 12 B.Subsequent foundational work on autoionization 13 VI.Continuing Story of P − P0 Spectroscopy for Zinc, Cadmium, and Mercury 14 VII.Continuing Story of Double Excitation in Helium 16 VIII.Autoionization as a pervasive effect in physics 18 Acknowledgments 19 References 19 Figures 22 I. -

Auger Cascades in Resonantly Excited Neon
Auger cascades in resonantly excited neon masterarbeit zur Erlangung des akademischen Grades „Master of Science“ eingereicht bei der Physikalisch-Astronomischen Fakultät der Friedrich-Schiller-Universität Jena von Sebastian Stock geboren am 21. März 1993 in Aalen Jena, März 2017 Die in dieser Arbeit präsentierten Ergebnisse wurden ebenfalls in dem folgenden Artikel veröentlicht: S. Stock, R. Beerwerth, and S. Fritzsche. “Auger cascades in resonantly excited neon”. To be submitted. Erstgutachter: Prof. Dr. Stephan Fritzsche Zweitgutachter: Priv.-Doz. Dr. Andrey Volotka Abstract ¿e Auger cascades following the resonant 1s → 3p and 1s → 4p excitation of neutral neon are studied theoretically. In order to accurately predict Auger electron spectra, shake probabilities, ion yields, and the population of nal states, the complete cascade of decays from neutral to doubly-ionized neon is simulated by means of extensive mcdf calculations. Experimentally known values for the energy levels of neutral, singly and doubly ionized neon are utilized in order to further improve the simulated spectra. ¿e obtained results are compared to experimental ndings. For the most part, quite good agreement between theory and experiment is found. However, for the lifetime widths of certain energy levels of Ne+, larger dierences between the calculated values and the experiment are found. It is presumed that these discrepancies originate from the approximations that are utilized in the calculations of the Auger amplitudes. Contents 1 Introduction1 2 Overview of the Auger cascades3 3 Theory 7 3.1 Calculation of Auger amplitudes........................7 3.2 ¿e mcdf method.................................8 3.3 Shake processes and the biorthonormal transformation...........9 4 Calculations 11 4.1 Bound state wave function generation.................... -

Chapter 1 Chemistry of Non-Aqueous Solutions
EFOP-3.4.3-16-2016-00014 projekt Lecture notes in English for the Chemistry of non-aqueous solutions, melts and extremely concentrated aqueous solutions (code of the course KMN131E-1) Pál Sipos University of Szeged, Faculty of Science and Informatics Institute of Chemistry Department of Inorganic and Analytical Chemistry Szeged, 2020. Cím: 6720 Szeged, Dugonics tér 13. www.u-szeged.hu www.szechenyi2020.hu EFOP-3.4.3-16-2016-00014 projekt Content Course description – aims, outcomes and prior knowledge 5 1. Chemistry of non-aqueous solutions 6 1.1 Physical properties of the molecular liquids 10 1.2 Chemical properties of the molecular liquids – acceptor and donor numbers 18 1.2.1 DN scales 18 1.2.1 AN scales 19 1.3 Classification of the solvents according to Kolthoff 24 1.4 The effect of solvent properties on chemical reactions 27 1.5 Solvation and complexation of ions and electrolytes in non-aqueous solvents 29 1.5.1 The heat of dissolution 29 1.5.2 Solvation of ions, ion-solvent interactions 31 1.5.3 The structure of the solvated ions 35 1.5.4 The effect of solvents on the complex formation 37 1.5.5 Solvation of ions in solvent mixtures 39 1.5.6 The permittivity of solvents and the association of ions 42 1.5.7 The structure of the ion-pairs 46 1.6 Acid-base reactions in non-aqueous solvents 48 1.6.1 Acid-base reactions in amphiprotic solvents of high permittivity 50 1.6.2 Acid-base reactions in aprotic solvents of high permittivity 56 1.6.3 Acid-base reactions in amphiprotic solvents of low permittivity 61 1.6.4 Acid-base reactions in amphiprotic solvents of low permittivity 61 1.7 The pH scale in non-aqueous solvents 62 1.8 Acid-base titrations in non-aqueous solvents 66 1.9 Redox reactions in non-aqueous solutions 70 2 EFOP-3.4.3-16-2016-00014 projekt 1.7.1 Potential windows of non-aqueous solvents 74 1.10 Questions and problems 77 2. -

Angular Distribution of Autoionization and Auger Electrons Ejected by Electron Impact from Laser-Excited and Polarized Atoms
J. Phys. B: At. Mol. Opt. Phys. 30 (1997) 1269–1291. Printed in the UK PII: S0953-4075(97)76979-7 Angular distribution of autoionization and Auger electrons ejected by electron impact from laser-excited and polarized atoms V V Balashov, A N Grum-Grzhimailo and N M Kabachnik Institute of Nuclear Physics, Moscow State University, Moscow 119899, Russia Received 8 August 1996, in final form 8 November 1996 Abstract. A general formalism is developed for a description of the angular distribution of electrons ejected after the electron-impact excitation/ionization of the laser-excited and polarized atoms. Properties of the angular distributions are analysed in a general case as well as in some particular geometries of the experiment. Within the two-step approximation the formalism is applied to the resonant single and double ionization with ejection of autoionization and Auger electrons. As an example the angular distributions of autoionization electrons from laser- excited sodium atoms are calculated in the plane-wave and distorted-wave Born approximations. Angular correlations between autoionization and scattered electrons, which can be measured in a coincidence (e, 2e) experiment with polarized targets, are also analysed. 1. Introduction The study of angular distributions of electrons ejected from atoms in electron–atom collisions is a well developed field which gives important information on mechanisms of excitation and decay of atomic quasistationary states (Mehlhorn 1985, 1990). In the majority of the studies, both experimental and theoretical, the electron-impact excitation from the ground states of atoms was investigated. Ten years ago Balashov and Grum-Grzhimailo (1986) suggested the investigation of the electron-impact excitation of atomic autoionizing states in atoms excited by lasers. -

Ultrafast Resonant Interatomic Coulombic Decay Induced by Quantum Fluid Dynamics
PHYSICAL REVIEW X 11, 021011 (2021) Ultrafast Resonant Interatomic Coulombic Decay Induced by Quantum Fluid Dynamics A. C. LaForge ,1,2,* R. Michiels,2 Y. Ovcharenko ,3,§ A. Ngai,2 J. M. Escartín ,4 N. Berrah ,1 C. Callegari ,5 A. Clark,6 M. Coreno,7 R. Cucini,5 M. Di Fraia,5 M. Drabbels,6 E. Fasshauer,8 P. Finetti,5 L. Giannessi,5,9 C. Grazioli ,18 D. Iablonskyi,10 B. Langbehn ,3 T. Nishiyama,11 V. Oliver,6 P. Piseri,12 O. Plekan,5 K. C. Prince ,5 D. Rupp,3,¶ S. Stranges ,13 K. Ueda,10 N. Sisourat,14 J. Eloranta,15 M. Pi,16,17 M. Barranco,16,17 F. Stienkemeier ,2 † ‡ T. Möller,3, and M. Mudrich8, 1Department of Physics, University of Connecticut, Storrs, Connecticut 06269, USA 2Institute of Physics, University of Freiburg, 79104 Freiburg, Germany 3Institut für Optik und Atomare Physik, Technische Universität Berlin, 10623 Berlin, Germany 4Institut de Química Teòrica i Computacional, Universitat de Barcelona, Carrer de Martí i Franqu`es 1, 08028 Barcelona, Spain 5Elettra-Sincrotrone Trieste, 34149 Basovizza, Trieste, Italy 6Laboratoire Chimie Physique Mol´eculaire, Ecole Polytechnique F´ed´erale de Lausanne, 1015 Lausanne, Switzerland 7CNR, Istituto di Struttura della Materia, 00016 Monterotondo Scalo, Italy 8Department of Physics and Astronomy, Aarhus University, 8000 Aarhus C, Denmark 9Nazionale di Fisica Nucleare, Laboratori Nazionali di Frascati, Via E. Fermi 40, 00044 Frascati, Roma, Italy CNR-Istituto Officina dei Materiali, Laboratorio TASC, 34149 Trieste, Italy 10Institute of Multidisciplinary Research for Advanced Materials, -
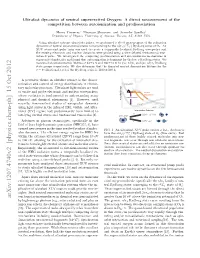
Ultrafast Dynamics of Neutral Superexcited Oxygen: a Direct Measurement of the Competition Between Autoionization and Predissociation
Ultrafast dynamics of neutral superexcited Oxygen: A direct measurement of the competition between autoionization and predissociation Henry Timmers,∗ Niranjan Shivaram, and Arvinder Sandhuy Department of Physics, University of Arizona, Tucson, AZ, 85721 USA. Using ultrafast extreme ultraviolet pulses, we performed a direct measurement of the relaxation 4 − dynamics of neutral superexcited states corresponding to the nlσg(c Σu ) Rydberg series of O2. An XUV attosecond pulse train was used to create a temporally localized Rydberg wavepacket and the ensuing electronic and nuclear dynamics were probed using a time-delayed femtosecond near- infrared pulse. We investigated the competing predissociation and autoionization mechanisms in superexcited molecules and found that autoionization is dominant for the low n Rydberg states. We measured an autoionization lifetime of 92±6 fs and 180±10 fs for (5s; 4d)σg and (6s; 5d)σg Rydberg state groups respectively. We also determine that the disputed neutral dissociation lifetime for the ν = 0 vibrational level of the Rydberg series is 1100 ± 100 fs. A pervasive theme in ultrafast science is the charac- terization and control of energy distributions in elemen- tary molecular processes. Ultrashort light pulses are used Energy to excite and probe electronic and nuclear wavepackets, Neutral whose evolution is fundamental to understanding many Dissociation Autoionization physical and chemical phenomena [1]. However, until Potential recently, time-resolved studies of wavepacket dynamics using light pulses in the infrared (IR), visible, and ultra- violet (UV) regime had predominantly been limited to Internuclear Distance low-lying excited states and femtosecond timescales [2]. Advances in photon technologies, specifically in the ( )+ field of laser high-harmonic generation (HHG)[3, 4], have opened new avenues in the time-resolved studies of molec- FIG. -

As Non-Aqueous Solvent
OPEN ACCESS Int. Res. J. of Science & Engineering, 2014, Vol. 2(1):1:18 ISSN: 2322-0015 REVIEW ARTICLE Dibromoacetic Acid: As Non-Aqueous Solvent Talwar Dinesh Chemistry Department, D.A.V. College, Chandigarh, India ABSTRACT KEYWORDS Water on account of several unique properties has been used as an ideal solvent for ionic Dibromoacetic compounds in the last three decades. This was the main reason that the solvents other than water acid, took so long to develop. It has been only since the late twenties that solution chemistry in non- non-aqueous aqueous solvents has been recognized as a separate and specialized branch of chemistry. Reactions solvent, of compounds susceptible to hydrolytic attack and even some routine chemical synthesis are now being extensively carried out in non-aqueous media. A large number of non-aqueous solvents have solvolytic been successfully explored and amongst them, mention may be made of such solvents as liquid reactions, ammonia, liquid sulphur dioxide, acetic anhydride, methyl alcohol etc. Acid base reactions have redox reactions, been carried out in ethylacetate, methyl formate and ethylchloroformate to explain the role played electrolytic by auto-ionization of the solvent. Mixed solvents are also used frequently in acid-base titrations and solvent, also in studying solvent-solute interactions. A systematic study of electrolytic solvent using mixed organic solvent organic solvents has been carried out. Acetic anhydride in mixture with nitromethane, acetonitrile, acetic acid and chloroform has been conveniently used for the estimation of weak bases. © 2013| Published by IRJSE INTRODUCTION references concerning to the use of these compounds in synthetic organic and inorganic chemistry (Macgookin, 1951). -

Probing and Controlling Autoionization Dynamics of Atoms with Attosecond Light Pulses
Probing and controlling autoionization dynamics of atoms with attosecond light pulses Wei-Chun Chu and C. D. Lin Abstract The time evolution of an autoionizing atomic system is studied theoreti- cally in the presence of a moderately intense dressing laser pulse. We first examine how an autoionizing wave packet evolves in time in the absence of an external field, and take the single 2pns(1P) resonances in beryllium as examples. Alternatively, we study the electron dynamics where an attosecond extreme ultraviolet (XUV) pulse excites two autoionizing states in the presence of a strong time-delayed coupling infrared (IR) laser pulse. The IR can be viewed as a probe to extract or a con- trol to modify the autoionization dynamics. The photoelectron and photoabsorption spectra are calculated for various time delays between the XUV and the IR pulses, and the results are compared with the available experiments. Finally, simulation of the coupled 2s2p(1P) and 2s2(1S) resonances in helium shows substantial spectral modifications by the dressing field parameters. Its analogy to electromagnetically induced transparency in the time domain is discussed. 1 Introduction The recent developments in attosecond light pulses have brought numerous appli- cations in measuring and controlling electron dynamics in the past decade [1, 2, 3]. Single attosecond pulses (SAPs), first realized in 2001 [4], have been used in the ex- periments in atomic [5, 6, 7], molecular [8, 9], and condensed systems [10]. These pulses, produced by the high-order harmonic generation (HHG) process of atoms in an intense infrared (IR) light source usually cover up to the extreme ultraviolet Wei-Chun Chu J. -

Photoelectron and Fragmentation Dynamics of the ${\Rm H}^{+} + {\Rm
PHYSICAL REVIEW RESEARCH 2, 043056 (2020) Photoelectron and fragmentation dynamics of the H+ + H+ dissociative channel in NH3 following direct single-photon double ionization Kirk A. Larsen,1,2,* Thomas N. Rescigno,2,† Travis Severt,3 Zachary L. Streeter,2,4 Wael Iskandar ,2 Saijoscha Heck ,2,5,6 Averell Gatton,2,7 Elio G. Champenois,1,2 Richard Strom,2,7 Bethany Jochim,3 Dylan Reedy,8 Demitri Call,8 Robert Moshammer,5 Reinhard Dörner ,6 Allen L. Landers,7 Joshua B. Williams,8 C. William McCurdy,2,4 Robert R. Lucchese,2 Itzik Ben-Itzhak ,3 Daniel S. Slaughter ,2 and Thorsten Weber 2,‡ 1Graduate Group in Applied Science and Technology, University of California, Berkeley, California 94720, USA 2Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA 3 J. R. Macdonald Laboratory, Physics Department, Kansas State University, Manhattan, Kansas 66506, USA 4Department of Chemistry, University of California, Davis, California 95616, USA 5Max-Planck-Institut für Kernphysik, Saupfercheckweg 1, 69117 Heidelberg, Germany 6J. W. Goethe Universität, Institut für Kernphysik, Max-von-Laue-Strasse 1, 60438 Frankfurt, Germany 7Department of Physics, Auburn University, Alabama 36849, USA 8Department of Physics, University of Nevada Reno, Reno, Nevada 89557, USA (Received 29 June 2020; accepted 21 September 2020; published 9 October 2020) We report measurements on the H+ + H+ fragmentation channel following direct single-photon double ionization of neutral NH3 at 61.5 eV, where the two photoelectrons and two protons are measured in coincidence using three-dimensional (3D) momentum imaging. We identify four dication electronic states that contribute to H+ + H+ dissociation, based on our multireference configuration-interaction calculations of the dication potential energy surfaces. -

Radiation & Autoionization Processes
Radiation & Autoionization Processes Atomic Theory and Computations | Lecture script | IAEA-ICTP School, Trieste, February 2017 Stephan Fritzsche Helmholtz-Institut Jena & Theoretisch-Physikalisches Institut, Friedrich-Schiller-Universit¨atJena, Fr¨obelstieg 3, D-07743 Jena, Germany Monday 27th February, 2017 1. Atomic theory and computations in a nut-shell 1.1. Atomic spectroscopy: Structure & collisions Atomic processes & interactions: ã Spontaneous emission/fluorescence: ... occurs without an ambient electromagnetic field; related also to absorption. ã Stimulated emission: ... leads to photons with basically the same phase, frequency, polarization, and direction of propagation as the incident photons. ã Photoionization: ... release of free electrons. ã Rayleigh and Compton scattering: ... Elastic and inelastic scattering of X-rays and gamma rays by atoms and molecules. ã Thomson scattering: ... elastic scattering of electromagnetic radiation by a free charged particle (electrons, muons, ions); low-energy limit of Compton scattering. ã Multi-photon excitation, ionization and decay: ... non-linear electron-photon interaction. ã Autoionization: ... nonradiative electron emission from (inner-shell) excited atoms. ã Electron-impact excitation & ionization: ... excited and ionized atoms; occurs frequently in astro-physical and laboratory plasmas. 3 1. Atomic theory and computations in a nut-shell ã Elastic & inelastic electron scattering: ... reveals electronic structure of atoms and ions; important for plasma physics. ã Pair production: ... creation of particles and antiparticles from the internal of light with matter (electron-positron pairs). ã Delbr¨uck scattering: ... deflection of high-energy photons in the Coulomb field of atomic nuclei; a consequence of vacuum polarization. ã ... ã In practice, the distinction and discussion of different atomic and electron-photon interaction processes also depends on the particular community/spectroscopy. -

Inorganic Chemistry
Inorganic Chemistry Acids, Bases and Non-Aqueous Solvents Prof. K N Upadhya 326 SFS Flats Ashok Vihar Phase IV Delhi -110052 CONTENTS Introduction Arrhenius Concept Bronsted-Lowry Concept of Acids and Bases Solvent-System Concept Aprotic Acids and Bases Lux-Flood Acid-Base Concept Strength of Bronsted Acids and Bases Relative Strengths of Acids and Bases Solvent Levelling and Discrimination in Water Amphoterism Trends in Acid Strength of various Bronsted Acids Acid Strength and Molecular Structure Strength of Mononuclear Oxoacids- Pauling’s Rules Lewis Acids and Bases Hard and Soft Acids and Bases Autoionization of Solvents Reactions in Non-Aqueous Solvents Discrimination in Non-Aqueous Solvents Keywords Arrhenius theory, Bronsted-Lowry theory, Solvent-system concept 1 Introduction The terms ‘acids’ and ‘bases’ have been defined in many ways. According to Arrhenius, probably the oldest, acids, and bases are the sources of H+ and OH- ions respectively. A somewhat broader but closely related definition (Bronsted – Lowry) is that an acid is a substance that supplies protons and a base is proton acceptor. Thus in water an acid increases the concentration of hydrated proton (H3O+) and a base lowers it or increases the concentration of OH-. In addition to Bronsted-Lowry concept there are others like solvent- solvent definition and the Lux and Flood definition but each one has its own limitations. One of the most general and useful of all definitions in reference to complex formations was due to G. N. Lewis. Lewis defined acid-base in terms of electron pair donor-acceptor capability. This definition includes Bronsted- Lowry definition as a special case. -

S Quantum Nuclear Dynamics in the Electron Transfer Mediated Decay Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Signature of the neighbor's quantum nuclear dynamics in the electron transfer mediated decay Cite this: Chem. Sci.,2021,12,9379 † All publication charges for this article spectra have been paid for by the Royal Society ab a a of Chemistry Aryya Ghosh, Lorenz S. Cederbaum * and Kirill Gokhberg We computed fully quantum nuclear dynamics, which accompanies electron transfer mediated decay (ETMD) in weakly bound polyatomic clusters. We considered two HeLi2 clusters – with Li2 being either in the singlet electronic ground state or in the triplet first excited state – in which ETMD takes place after + ionization of He. The electron transfer from Li2 to He leads to the emission of another electron from Li2 into the continuum. Due to the weak binding of He to Li2 in the initial states of both clusters, the involved nuclear wavepackets are very extended. This makes both the calculation of their evolution and the interpretation of the results difficult. We showed that despite the highly delocalized nature of the Received 12th March 2021 wavepackets the nuclear dynamics in the decaying state is imprinted on the ETMD electron spectra. The Accepted 1st June 2021 Creative Commons Attribution-NonCommercial 3.0 Unported Licence. analysis of the latter helps understanding the effect which electronic structure and binding strength in DOI: 10.1039/d1sc01478a the cluster produce on the quantum motion of the nuclei in the decaying state. The results produce rsc.li/chemical-science a detailed picture of this important charge transfer process in polyatomic systems.