A Dormant Microbial Component in the Development of Pre-Eclampsia1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Complete Issue
Conerete Bloek & Sprayed Coating- a ~inning eOlDbination of beauty & silDplieity at lo~eost STAND RD CREGO CK WITH CEMENT LE E COAT AND SPRAYED-ON TE U ED INISH COAT • JERRY GOFFE PHOTO RUST TRACTOR COMPANY ELLISON-HAWKINS-VOGT 6' BYRNES, P.A. ARCH ITECTS ·ENGINEERS K. L. HOUSE CONSTRUCTION CO. GENERAL CONTRACTOR KENNETH P. THOMPSON CO., INC. MASONRY CONTRACTOR BILL C. CARROLL CO., INC. SPRAYED COATING "'\ For our reoders I"~, we wish 1976 to \'\'~" be rhapsodic, • thriving, abundant and eudaemonic. , '~-I4 ""_ """""' .----"''''''v~J col: 18 no. 1 jan. - feb. 1976 • new mexico architecture As we begin another year of New Mexico Architecture, it is appropri ate to remind our readers of the contribution made to our financial stability by the advertisers. It is their support which makes possible the production of the magazine. To all of these fine people the "staff" says a most sincere thank you! Space in New Mexico Architecture o DOD as a Resource for on Energy Ethic 10 Beginning on page lOis an arti - By Anthony C. Antoniades, AI.A, AI.P. cle by Anthony C. Antoniades, AlA, AlP, Associate Professor of Archi tecture at the University of Texas at Arlington. Professor Antoniades taught architecture at the Universi ty of New Mexico before moving to Index to Advertisers 18 Texas. It was during those years in our state that he developed a strong interest in and knowledge of the architectural heritage of New Mexi ico. Three articles by Antoniades hove appeared previously in NMA November/December 1971, Septem ber/October 1973 and July/August 1974. -

Annual Report
Greeks Helping Greeks ANNUAL REPORT 2019 About THI The Hellenic Initiative (THI) is a global, nonprofi t, secular institution mobilizing the Greek Diaspora and Philhellene community to support sustainable economic recovery and renewal for Greece and its people. Our programs address crisis relief through strong nonprofi t organizations, led by heroic Greeks that are serving their country. They also build capacity in a new generation of heroes, the business leaders and entrepreneurs with the skills and values to promote the long term growth of Hellas. THI Vision / Mission Statement Investing in the future of Greece through direct philanthropy and economic revitalization. We empower people to provide crisis relief, encourage entrepreneurs, and create jobs. We are The Hellenic Initiative (THI) – a global movement of the Greek Diaspora About the Cover Featuring the faces of our ReGeneration Interns. We, the members of the Executive Committee and the Board of Directors, wish to express to all of you, the supporters and friends of The Hellenic Initiative, our deepest gratitude for the trust and support you have given to our organization for the past seven years. Our mission is simple, to connect the Diaspora with Greece in ways which are valuable for Greece, and valuable for the Diaspora. One of the programs you will read about in this report is THI’s ReGeneration Program. In just 5 years since we launched ReGeneration, with the support of the Coca-Cola Co. and the Coca-Cola Foundation and 400 hiring partners, we have put over 1100 people to work in permanent well-paying jobs in Greece. -

TRUST, but VERIFY: WHY the BLOCKCHAIN NEEDS the LAW Kevin Werbach†
TRUST, BUT VERIFY: WHY THE BLOCKCHAIN NEEDS THE LAW Kevin Werbach† ABSTRACT The blockchain could be the most consequential development in information technology since the Internet. Created to support the Bitcoin digital currency, the blockchain is actually something deeper: a novel solution to the age-old human problem of trust. Its potential is extraordinary. Yet, this approach may not promote trust at all without effective governance. Wholly divorced from legal enforcement, blockchain-based systems may be counterproductive or even dangerous. And they are less insulated from the law’s reach than it seems. The central question is not how to regulate blockchains but how blockchains regulate. They may supplement, complement, or substitute for legal enforcement. Excessive or premature application of rigid legal obligations will stymie innovation and forego opportunities to leverage technology to achieve public policy objectives. Blockchain developers and legal institutions can work together. Each must recognize the unique affordances of the other system. DOI: https://doi.org/10.15779/Z38H41JM9N © 2018 Kevin Werbach. † Associate Professor of Legal Studies and Business Ethics, The Wharton School, University of Pennsylvania. Email: [email protected]. Thanks to Dan Hunter for collaborating to develop the ideas that gave rise to this Article, and to Sarah Light, Patrick Murck, and participants in the 2017 Lastowka Cyberlaw Colloquium and 2016 TPRC Conference for comments on earlier drafts. 488 BERKELEY TECHNOLOGY LAW JOURNAL [Vol. 33:487 -

Sampling and Quantification of Biofilms in Food Processing And
21 Sampling and quantifi cation of biofi lms in food processing and other environments D. E. Nivens, and B. M. Co, Purdue University, USA and M. J. Franklin, The Center for Biofi lm Engineering, USA Abstract: In the food industry, assessment of food contact surfaces is necessary to determine whether equipment is properly cleaned and/or sanitized and whether living problematic microorganisms are present. Existing quantitative detection technologies are limited by the inability to directly detect living cells in sporadically dispersed biofi lms on large surface areas. Thus, precise and accurate sampling strategies must be coupled with detection technology. This chapter discusses sampling methods and standard (e.g., plating and ATP-bioluminescence) and emerging (e.g., spectrometry, immunosensor, and nucleic acid-based) quantitative techniques to detect biofi lms on food contact surfaces with a survey of function, analytical performance, and limitations. Key words: biofi lm, sampling methods, spectrometry, immunosensors, nucleic acid-based. 21.1 Introduction Defi nitions of biofi lms vary but most investigators agree that biofi lms usually consist of four main components: (i) water, (ii) the living microbial population, (iii) the surface to which the cells are adhered, and (iv) the associated extracellular matrix, which can consist of secreted polymers (2, 17, 20, 107). These polymers, usually polysaccharides and/or protein, allow the cells to form three-dimensional structures but are not required for biofi lms to proliferate on a surface (79). Biofi -

CHSAA New York State Intersectional Championships
Icahn Stadium - Site License Hy-Tek's MEET MANAGER Page 1 2017 CHSAA Intersectional Championship - 5/27/2017 Icahn Stadium Results 53 Kelleher, Maggie Notre Dame A 15.67 0.5 Girls 100 Meter Dash Red 54 Gaston, Edjinanie St. John's P 15.74 NWI Name School Prelims 55 Bonner, Brianna Cardinal Spe 15.76 1.0 Preliminaries 56 Keorner, Mia Maria Regina 15.78 1.4 1 Hazzard, Halle St. Anthony' 11.86q 0.5 57 Llamoza, Isabella St. Agnes 15.84 NWI 2 Green, Zhanna St. John the 12.42q 1.1 58 Barretta, Cristina St. Francis 16.12 1.4 3 Davis, Tayla Cardinal Spe 12.72q 1.6 59 Polanco, Melanie St. Agnes 16.28 1.4 4 William, Shemayah Nazareth 12.88q 1.4 60 Nakervis, Veronica St. Anthony' 16.31 1.1 5 Connolly, Jayme Our Lady of 13.04q 1.7 61 Guaman, Odalis Archbishop M 16.42 1.7 6 Tang, Amy St. Anthony' 13.08q 1.4 62 Deck, Victoria St. Francis 16.53 NWI 7 McKenzie, Garcelle St. Joseph's 13.20q 1.6 63 Voltus, Jomara Archbishop M 16.68 1.1 8 Monteforte, Samantha St. Joseph b 13.21q 1.0 64 Luna, Jennifer St. Agnes 16.94 1.7 9 Hufford, Erin St. Mary's L 13.28 NWI 65 Vasquez, Alanah Cardinal Spe 17.39 1.4 10 Szanyi, Lauren Nardin Acade 13.29 NWI 66 Mendoza, Destiny Mary Louis A 17.68 1.1 11 Harrington, Alyssa Cardinal O'H 13.33 1.4 67 Francis, Ayanna Mary Louis A 17.90 1.4 12 Charles, Amanda St. -

2009 Paris, France the Movement Disorder Society’S 13Th International Congress of Parkinson’S Disease and Movement Disorders
FINAL PROGRAM The Movement Disorder Society’s 13th International Congress OF PARKINSon’S DISEASE AND MOVEMENT DISORDERS JUNE 7-11, 2009 Paris, France The Movement Disorder Society’s 13th International Congress of Parkinson’s Disease and Movement Disorders Claiming CME Credit To claim CME credit for your participation in the MDS 13th International Congress of Parkinson’s Disease and Movement Disorders, International Congress participants must complete and submit an online CME Request Form. This Form will be available beginning June 10. Instructions for claiming credit: • After June 10, visit www.movementdisorders.org/congress/congress09/cme • Log in following the instructions on the page. You will need your International Congress Reference Number, located on the upper right of the Confirmation Sheet found in your registration packet. • Follow the on-screen instructions to claim CME Credit for the sessions you attended. • You may print your certificate from your home or office, or save it as a PDF for your records. Continuing Medical Education The Movement Disorder Society is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians. Credit Designation The Movement Disorder Society designates this educational activity for a maximum of 30.5 AMA PRA Category 1 Credits™. Physicians should only claim credit commensurate with the extent of their participation in the activity. Non-CME Certificates of Attendance were included with your on- site registration packet. If you did not receive one, please e-mail [email protected] to request one. The Movement Disorder Society has sought accreditation from the European Accreditation Council for Continuing Medical Education (EACCME) to provide the following CME activity for medical specialists. -

Kalamazoo County Naturalization
Kalamazoo County Naturalization Last Name First Name Middle Name First Paper Second Paper Aach Rita Ann V66P5111 Aalbregtse Abraham Peter V10P163 Aalbregtse Johannes V12P446 Aalbregtse Jozias V11P151 Aalbregtse Suzanna Maria V58P3802 Aarssen Diederika Frederika Willemina V13P40 Abbate Santi V13P56 Abbate Santi V34P29 V34P29 Abolins Aina V64P4686 Abolins Augusts V64P4687 Abolins Guntis V64P4688 Abolins Mara V64P4627 Aboshin Aglou Ismail V29P64 V29P64 Abraham Francis T V10P219 Abraham Francis Thurlborn V23P104 V23P104 Abraham Maurice George V10P237 Abraham Thomas J V4P41 Abraham Thomas J V5P134 Abrahamse Pieter V3P262 Abromson Isral V5P314 Achterhof Albert V64P4603 Achterhof Johanna V59P3900 Adam Ben V52P3134 Adam Enke V14P204 Adam Simon V11P102 Adam Simon V14P213 Friday, July 06, 2007 Page 1 of 577 Kalamazoo County Naturalization Index Order copies of records by calling (517) 373-1408 Archives of Michigan Home page: www.michigan.gov/archivesofmi E-mail: [email protected] Last Name First Name Middle Name First Paper Second Paper Adam Simon V35P8 V35P8 Adamopooulos Fotis V34P39 V34P39 Adamopoulos Fotis V14P316 Adamopoulos James V30P27 Adams Aaltise V19P3868 Adams Antje V17P3172 Adams Claus John V51P3022 Adams Ella Nettie V71P27 Adams Enke V18P3596 Adams Frank V14P316 Adams Frank V34P39 V34P39 Adams Jacoba V57P3649 Adams James V30P27 Adams Kaert V46P2556 Adams Koert V46P2556 Adams Magdalena Anne V51P3034 Adams Nicky V58P3833 Adkin Marion Jean V53P3215 Adler Nathan V52P3189 Advocaat John Edward V21P4397 Aelick William John V23P142 V23P142 -
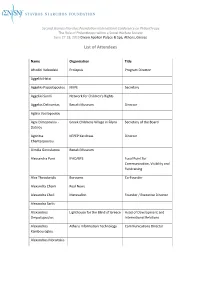
List of Attendees
Second Stavros Niarchos Foundation International Conference on Philanthropy The Role of Philanthropy within a Social Welfare Society June 27-28, 2013 Divani Apollon Palace & Spa, Athens, Greece List of Attendees Name Organization Title Afroditi Veloudaki Prolepsis Program Director Aggeliki Hatzi Aggeliki Papadopoulou KIKPE Secretary Aggeliki Sandi Network for Children's Rights Aggelos Delivorrias Benaki Museum Director Aglaia Vasilopoulou Agni Dimopoulou - Greek Childrens Village in Filyro Secretary of the Board Datsiou Agoritsa KEPEP Karditsas Director Chantzopoulou Aimilia Geroulanou Benaki Museum Alessandra Pani IFAD/BFS Focal Point for Communication, Visibility and Fundraising Alex Theodoridis Boroume Co-Founder Alexandra Chaini Real News Alexandra Choli Metavallon Founder / Executive Director Alexandra Sarlis Alexandros Lighthouse for the Blind of Greece Head of Development and Despotopoulos International Relations Alexandros Athens Information Technology Communications Director Kambouroglou Alexandros Moraitakis Name Organization Title Alexandros Taxildaris Association for People with President Mobility Problems and Friends Perpato Alexia Divani Alexia Kotsopoulou AWOG Representative Alexia Raphael Stavros Niarchos Foundation Intern Aliki Martinou Mazigia to Paidi Aliki Mitsakou Aliki Tserketzoglou Galilee Palliative Care Unit Amalia Delicari Stavros Niarchos Foundation Associate Program Officer Amalia Zeppou Municipality of Athens Amvrosios Holy Metropolis of Kalavryta and Metropolitan Bishop Aegialia Anastasia Andritsou British -

Oss Personnel Files - Rg 226 Entry 224 12/23/2010
OSS PERSONNEL FILES - RG 226 ENTRY 224 12/23/2010 LAST NAME FIRST NAME M I RANK or SERIAL BOX NOTES LOCATION Aaberg Caf-3 E T-5 37541999 1 230/86/26/03 Aalbu Sigurd J S/Sgt 37091702 1 230/86/26/03 Aanonsen Olaf H Cpl 32440677 1 230/86/26/03 Abbenante Helen G Cpl A125357 1 230/86/26/03 Abbote Frank Pfc 32985690 1 230/86/26/03 Abbote John A T-3 42048684 1 230/86/26/03 Abbott Delbert H Pvt 35314960 1 230/86/26/03 Abbott Floyd H Pfc 39535381 1 230/86/26/03 Abbott Frederick K Caf-11 1 230/86/26/03 Abbott James E S/Sgt 12063651 1 230/86/26/03 Abbott James F T-5 11102233 1 230/86/26/03 Abbott Norman SP 2/c 5634486 1 230/86/26/03 Abbott Robert J Sgt 12140436 1 230/86/26/03 Abbott Victor J M/Sgt 32455694 1 230/86/26/03 Abee Robert V T-4 34437404 1 230/86/26/03 Abel Arthur A 1st Lt 1181596 1 230/86/26/03 Abel Calvin J T-5 16117938 1 230/86/26/03 Abel John C 1 230/86/26/03 Abele Charles R 1st Lt 462544 1 230/86/26/03 Abele Herbert, Jr. A 2nd Lt 1329784 1 230/86/26/03 Abendschein Norman W Sgt 33191772 1 230/86/26/03 Abernathy James D 1st Lt 343733 1 230/86/26/03 Abney James K 1 230/86/26/03 Abraham Michael K Capt 1010362 1 230/86/26/03 Abrahamovitz Moses T/Sgt 33751543 2 230/86/26/03 Abrahams Isaace L 39090318 1 230/86/26/03 Abrahamson Albert Pvt 32966288 1 230/86/26/03 Abrahamson John D P-4 1 230/86/26/03 Abrams Allen P-7 2 230/86/26/03 Abrams Leonard 2 230/86/26/03 Abrams Melville F T-4 32978088 2 230/86/26/03 Abrams Ruth B Caf-7 2 230/86/26/03 Abrignani Vincent A Maj 1297132 2 230/86/26/03 Abrogast Hazel E Caf-4 2 230/86/26/03 Abromaitis Alexander -

2020 PRELIMINARY VALUES TOWN of MOULTONBOROUGH REPORT by OWNER's NAME Total Assessed Total Assessed Total Assessed Owner Parcel ID Location Land Improvements Value
2020 PRELIMINARY VALUES TOWN OF MOULTONBOROUGH REPORT BY OWNER'S NAME Total Assessed Total Assessed Total Assessed Owner Parcel ID Location Land Improvements Value 1 FIELDSTONE WAY REALTY TRUST 000024 / 004 / 001 / 000 / 000 FIELDSTONE WAY 135,500 0 135,500 10 SECOND POINT REALTY TRUST 000133 / 039 / 000 / 000 / 000 10 SECOND POINT ROAD 630,300 589,800 1,220,100 100 SERIES SEWER SYS ASSOC 000174 / 075 / 000 / 000 / 000 KRAINEWOOD DRIVE 0 0 0 103 EVANS ROAD NOMINEE TRUST 000005 / 008 / 000 / 000 / 000 EVANS ROAD 62,300 0 62,300 103 EVANS ROAD NOMINEE TRUST 000005 / 007 / 000 / 000 / 000 103 EVANS ROAD 83,500 169,400 252,900 1040 WHITTIER LLC 000043 / 018 / 000 / 000 / 000 WHITTIER HIGHWAY 44,900 0 44,900 1040 WHITTIER LLC 000043 / 019 / 000 / 000 / 000 1040 WHITTIER HIGHWAY 97,800 407,200 505,000 111 KIMBALL DRIVE PROPERTY TRUST 000223 / 075 / 001 / 000 / 000 KIMBALL DRIVE 89,800 0 89,800 111 KIMBALL DRIVE PROPERTY TRUST 000223 / 045 / 000 / 000 / 000 111 KIMBALL DRIVE 616,500 391,600 1,008,100 113 EVANS ROAD REV TRUST 000005 / 009 / 000 / 000 / 000 113 EVANS ROAD 61,500 10,900 72,400 12 GANSY ISLAND MOULTONBOROUGH LLC 000130 / 067 / 000 / 000 / 000 12 GANSY ISLAND 177,900 239,200 417,100 123 KIMBALL DRIVE TRUST 000223 / 047 / 000 / 000 / 000 123 KIMBALL DRIVE 492,500 169,800 662,300 1241 WHITTIER HIGHWAY LLC 000018 / 017 / 000 / 000 / 000 1241 WHITTIER HIGHWAY 99,600 240,900 340,500 126 FAR ECHO ROAD REALTY TRUST 000245 / 020 / 000 / 000 / 000 126 FAR ECHO ROAD 158,900 99,100 258,000 128 LEE ROAD LLC 000068 / 001 / 000 / 000 / 000 LEE -

The One Hundred Twenty-Ninth Commencement of Saint Peter's University
The One Hundred Twenty-Ninth Commencement of Saint Peter ’s University Friday, May 21, 2021 Message from the President May 21, 2021 Dear Members of the Class of 2020: Today, we gather to recognize your achievement. I know this day looks a lot different from what you envisioned when you began your journey at Saint Peter’s. Your final weeks of being on-campus last year were abruptly uprooted. And yet during a time of great uncertainty you rose to the challenge, demonstrating great flexibility as you faced situations never faced before. While you have already begun the next chapter in your life, let us take this moment to officially recognize the successful completion of your degree. I am confident that your education has prepared you to excel intellectually, lead ethically, serve compassionately and promote justice as your journey continues. The Ignatian values instilled in you have provided a solid intellectual, moral and spiritual foundation on which to build a life full of gratitude, knowledge and community. Unquestionably, the Class of 2020 consists of future leaders who will forever be “men and women for others.” Know that as you join our Peacock family of more than 36,000 alumni, I am so proud of you and all that you have accomplished while here. You are part of a 129-year tradition and I look forward to seeing where the next journey takes you. In the words of Saint Ignatius, “go forth and set the world on fire.” Remember to stay in touch with alma mater so that we can continue to celebrate your successes. -
![Download/Report.Htm] World Commission on Dams, 2000: Dams and Development: a New Framework for Decision-Making](https://docslib.b-cdn.net/cover/3657/download-report-htm-world-commission-on-dams-2000-dams-and-development-a-new-framework-for-decision-making-1823657.webp)
Download/Report.Htm] World Commission on Dams, 2000: Dams and Development: a New Framework for Decision-Making
INTERGOVERNMENTAL PANEL ON CLIMATE CHANGE WMO UNEP _______________________________________________________________________________________________________________________ INTERGOVERNMENTAL PANEL IPCC-XXVIII/Doc.13 ON CLIMATE CHANGE (8.IV.2008) TWENTY-EIGHTH SESSION Agenda item: 10 Budapest, 9-10 April 2008 ENGLISH ONLY TECHNICAL PAPER ON CLIMATE CHANGE AND WATER (Finalized at the 37th Session of the IPCC Bureau) _______________________________________________________________________________________________________________________ IPCC Secretariat, c/o WMO, 7bis, Avenue de la Paix, C.P. N° 2300, 1211 Geneva 2, SWITZERLAND Phone: +41 22 730 8208/8254/8284 Fax: +41 22 730 8025/8013 E-mail: [email protected] Website: http://www.ipcc.ch INTERGOVERNMENTAL PANEL ON CLIMATE CHANGE WMO UNEP Climate Change and Water Bryson Bates, Zbigniew W. Kundzewicz, Shaohong Wu, Nigel Arnell, Virginia Burkett, Petra Döll, Daniel Gwary, Clair Hanson, BertJan Heij, Blanca Jiménez, Georg Kaser, Akio Kitoh, Sari Kovats, Pushpam Kumar, Chris Magadza, Daniel Martino, Luis José Mata, Mahmoud Medany, Kathleen Miller, Taikan Oki, Balgis Osman, Jean Palutikof, Terry Prowse, Roger Pulwarty, Jouni Räisänen, Jim Renwick, Francesco Tubiello, Richard Wood, Zong-Ci Zhao, Julie Arblaster, Richard Betts, Aiguo Dai, Christopher Milly, Linda Mortsch, Leonard Nurse, Richard Payne, Iwona Pinskwar, Tom Wilbanks SUBJECT TO FINAL COPY EDIT This is a Technical Paper of the Intergovernmental Panel on Climate Change prepared in response to a decision of the Panel. The material herein has undergone expert and government review, but has not been considered by the Panel for possible acceptance or approval. April 2008 This paper was prepared under the auspices of the IPCC, which is chaired by Dr. Rajendra K. Pachauri, and administered by the IPCC Working Group II Technical Support Unit.