Virtual Screening of C. Sativa Constituents for the Identification Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CBD (Cannabidiol)
TRANSPORTATION RESEARCH BOARD Driving Toward the Truth - Dispelling the Myths About Cannabis Products February 10, 2021 @NASEMTRB #TRBwebinar PDH Certification The Transportation Research Board has met the standards and Information: requirements of the Registered Continuing Education Providers •1.5 Professional Development Program. Credit earned on completion Hour (PDH) – see follow-up of this program will be reported to email for instructions RCEP. A certificate of completion will •You must attend the entire be issued to participants that have registered and attended the entire webinar to be eligible to receive session. As such, it does not include PDH credits content that may be deemed or •Questions? Contact Reggie construed to be an approval or Gillum at [email protected] endorsement by RCEP. #TRBwebinar Learning Objectives 1. Identify impacts of the Farm Bill on use of THC and CBD products 2. Describe the toxicology of THC and CBD products 3. Discuss how THC and CBD products affect driving performance and crash risk #TRBwebinar TRB Standing Committee on Impairment in Transportation (ACS50) TRB Webinar: Driving Toward the Truth - Dispelling the Myths About Cannabis Products Dr. Barry K. Logan Executive Director, Center for Forensic Science Research and Education (CFSRE); Senior Vice President of Forensic Sciences, and Chief Scientist at NMS Labs Michelle Peace, Ph.D. Associate Professor and PI, Laboratory for Forensic Toxicology Research Department of Forensic Science, Virginia Commonwealth University Dr. Darrin Grondel Vice President, -

What Is Delta-8 THC?? Cannabinoid Chemistry 101
What is Delta-8 THC?? Cannabinoid Chemistry 101 National Conference on Weights and Measures Annual Meeting - Rochester, NY Matthew D. Curran, Ph.D. July 21, 2021 Disclaimer Just to be clear… • I am a chemist and not a lawyer so: • This presentation will not discuss the legal aspects of Δ8-THC or DEA’s current position. • This presentation will not discuss whether Δ8-THC is considered “synthetic” or “naturally occurring.” • This is not a position statement on any issues before the NCWM. • Lastly, this should only be considered a scientific sharing exercise. Florida Department of Agriculture and Consumer Services 2 Cannabis in Florida Cannabis Syllabus • What is Cannabis? • “Mother” Cannabinoid • Decarboxylation • Relationship between CBD and THC • What does “Total” mean? • Dry Weight vs. Wet Weight • What does “Delta-9” mean? • Relationship between “Delta-8” and “Delta-9” • CBD to Delta-8 THC • Cannabinoid Chemistry 202… Florida Department of Agriculture and Consumer Services 3 Cannabis Cannabis • Cannabis sativa is the taxonomic name for the plant. • The concentration of Total Δ9-Tetrahydrocannabinol (Total Δ9-THC) is critical when considering the varieties of Cannabis sativa. • Hemp – (Total Δ9-THC) 0.3% or less • Not really a controversial term, “hemp” • Marijuana/cannabis – (Total Δ9-THC) Greater than 0.3% • Controversial term, “marijuana” • Some states prohibit the use of this term whereas some states have it in their laws. • Some states use the term “cannabis.” • Not italicized • Lower case “c” Florida Department of Agriculture and -

In Silico Assessment of Drug-Like Properties of Phytocannabinoids in Cannabis Sativa
EDUCATUM JSMT Vol. 4 No. 2 (2017) ISSN 2289-7070 / eISSN 2462-2451 (1-7) https://ejournal.upsi.edu.my/journal/EDSC In Silico Assessment of Drug-Like Properties of Phytocannabinoids in Cannabis Sativa Shakinaz Desa1*, Asiah Osman2, and Richard Hyslop3 1Department of Biology, Universiti Pendidikan Sultan Idris, Malaysia, 2Natural Product Division, Forest Research Institute Malaysia, 3Department of Chemistry and Biochemistry, University of Northern Colorado, USA *Corresponding author: [email protected] Abstract This study investigated drug-like properties of phytocannabinoids in Cannabis sativa using an in silico study. We report sixteen phytocannabinoids: cannabidiol (CBD), cannabidiolic acid (CBDA), cannabinol (CBN), cannabichromene (CBC), cannabigerol (CBG), cannabicyclol (CBL), cannabivarin (CBV), cannabidivarin (CBDV), cannabichromevarin (CBCV), cannabigerovarin (CBGV), cannabinodiol (CBDL), cannabielsoin (CBE), cannabitriol (CBT), Δ9-tetrahydrocannabinol (Δ9-THC), Δ9-tetrahydrocannabivarin (Δ9-THCV), and Δ8-tetrahydrocannabinol (Δ8-THC). All chemical structures and properties were obtained from PubChem Compound, National Center for Biotechnology Information, U.S. National Library of Medicine. Molinspiration was used for the calculation of molecular properties and bioactivity score. The parameters were molecular weight (MW), number of hydrogen acceptor (HBA), number of hydrogen donor (HBD), partition coefficient (cLogP), polar surface area (PSA) and number of rotatable bonds (NROTB). We predicted bioactivity scores for G Protein-Coupled Receptors (GPCR) ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor. Lipinski’s rule was used as reference to determine the drug-like properties of the phytocannabinoids. All compounds have MW<500, HBA<10, HBD<5, TPSA<140Å2 and NRTOB<10. Bioactivity score showed an active or moderately active in all compounds. -

(12) United States Patent (10) Patent No.: US 8.449,908 B2 Stinchcomb Et Al
USOO84499.08B2 (12) United States Patent (10) Patent No.: US 8.449,908 B2 Stinchcomb et al. (45) Date of Patent: May 28, 2013 (54) TRANSDERMAL DELIVERY OF 4,663,474 A 5, 1987 Urban CANNABIDOL 4,876,276 A 10, 1989 Mechoulam et al. 5,059,426 A * 10/1991 Chiang et al. ................. 424/449 5,223,262 A 6/1993 Kim et al. (75) Inventors: Audra L. Stinchcomb, Lexington, KY 5,254,346 A 10, 1993 Tucker et al. (US); Buchi N. Nalluri, Lexington, KY (US) (Continued) FOREIGN PATENT DOCUMENTS (73) Assignee: Alltranz, LLC, Lexington, KY (US) EP 0570920 A1 5, 1992 EP O656354 B1 12/1993 (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 (Continued) U.S.C. 154(b) by 1546 days. OTHER PUBLICATIONS (21) Appl. No.: 11/157,034 "Cannabinoid system as a potential target for drug development in the treatment of cardiovascular disease” by Mendizabal et al., Current (22) Filed: Jun. 20, 2005 Vascular Pharmacology, 2003, vol. 1, No. 3, 301-313.* (65) Prior Publication Data (Continued) US 2005/0266.061 A1 Dec. 1, 2005 Primary Examiner — Isis Ghali (74) Attorney, Agent, or Firm — Foley & Lardner LLP (51) Int. C. A6DF 3/00 (2006.01) (57) ABSTRACT A6DF 3/02 (2006.01) A 6LX 9/70 (2006.01) The present invention overcomes the problems associated A6IL 15/16 (2006.01) with existing drug delivery systems by delivering cannab U.S. C. inoids transdermally. Preferably, the cannabinoids are deliv (52) ered via an occlusive body (i.e., a patch) to alleviate harmful USPC ........................... -

The Phytocannabinoides from Cannabis Sativa L. an Overview
Hop and Medicinal Plants, Year XXVIII, No. 1-2, 2020 ISSN 2360–0179 print, ISSN 2360–0187 electronic THE PHYTOCANNABINOIDES FROM CANNABIS SATIVA L. AN OVERVIEW ONA Andreea Daniela, Sorin MUNTEAN, Leon MUNTEAN* University of Agricultural Sciences and Veterinary Medicine, Faculty of Agriculture, Crop Science Department 3-5 Mănăstur Street, 400372, Cluj-Napoca, Romania *Corresponding author e-mail: [email protected] Abstract: Cannabis sativa is among the first cultivated plants for producing fibres from the stalks and consumption of the seeds. Later, the medicinal properties were discovered. The oldest written record of using cannabis for the medicinal and recreational purposes is from China. Over the time many experimental researches were carried out that emphasize the importance of cannabinoids for human health. The present paper focused to present an overview of the main cannabinoids synthetized in cannabis plant and their medicinal and therapeutic effects. Cannabis has a complex chemical composition including besides cannabinoids (over 100), also terpenoids, sugars, alkaloids, quinones and stilbenoids. Of major importance are two compounds tetrahydrocannabinol (THC) and cannabidiol (CBD). Over the time different researches pointed out the beneficial effect of cannabinoids on a wide range of diseases such as epilepsy, neurological diseases, mental conditions like anxiety and depression, chronic pains and certain forms of cancer. In addition to the important advantages it must be also paid attention to the psychoactive action of the certain cannabinoids that may produce cognitive disturbances, attention disorders, sever panic, can affect the short-term memory, and slow the reaction time. This deficits induced usually by THC can be moderate by using CBD which reduce the psychoactive effect. -
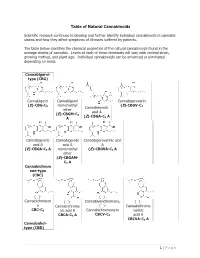
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic -

Cannabinoids and Zebrafish Issue Date: 2013-05-22
Cover Page The handle http://hdl.handle.net/1887/20899 holds various files of this Leiden University dissertation. Author: Akhtar, Muhammad Tayyab Title: Cannabinoids and zebrafish Issue Date: 2013-05-22 Cannabinoids and zebrafish Muhammad Tayyab Akhtar Muhammad Tayyab Akhtar Cannabinoids and zebrafish ISBN: 978-94-6203-345-0 Printed by: Wöhrmann Print Service Cover art and designed by M khurshid and MT Akhtar Cannabinoids and zebrafish PROEFSCHRIFT ter verkrijging van de graad van Doctor aan de Universiteit Leiden, op gezag van Rector Magnificus prof.mr. C.J.J.M. Stolker, volgens besluit van het College voor Promoties te verdedigen op woensdag 22 mei 2013 klokke 10:00 uur door Muhammad Tayyab Akhtar geboren te Rahim Yar Khan (Pakistan) in 1984 Promotiecommissie Promotor: Prof. Dr. R. Verpoorte Co-promotores: Dr. F. van der Kooy Dr. Y.H. Choi Overige leden: Prof. Dr. S. Gibbons (The School of Pharmacy, London) Dr. F. Hollmann (TU Delft) Prof. Dr. M.K. Richardson Prof. Dr. P.G.L. Klinkhamer Prof. Dr. C.J. ten Cate To My Father and Family! CONTENTS Chapter 1 General introduction 9 Chapter 2 Biotransformation of cannabinoids 21 Chapter 3 Hydroxylation and further oxidation of Δ9-tetrahydrocannabinol by alkane-degrading bacteria. 53 Chapter 4 Hydroxylation and glycosylation of Δ9-THC by Catharanthus roseus cell suspension culture analyzed by HPLC-PDA and mass spectrometry 73 Chapter 5 Developmental effects of cannabinoids on zebrafish larvae 89 Chapter 6 Metabolic effects of cannabinoids in zebrafish (Danio rerio) embryo determined by 1H NMR metabolomics. 117 Chapter 7 Metabolic effects of carrier solvents and culture buffers in zebrafish embryos determined by 1H NMR metabolomics. -

Vermont Hemp Rules
FINAL PROPOSED RULE # Administrative Procedures— Final Proposed Rule Filing Instructions: In accordance with Title 3 Chapter 25 of the Vermont Statutes Annotated and the "Rule on Rulemaking" adopted by the Office of the Secretary of State, this filing will be considered complete upon filing and acceptance of these forms with the Office of the Secretary of State, and the Legislative Committee on Administrative Rules. All forms requiring a signature shall be original signatures of the appropriate adopting authority or authorized person, and all filings are to be submitted at the Office of the Secretary of State, no later than 3:30 pm on the last scheduled day of the work week. The data provided in text areas of these forms will be used to generate a notice of rulemalcing in the portal of "Proposed Rule Postings" online, and the newspapers of record if the rule is marked for publication. Publication of notices will be charged back to the promulgating agency. PLEASE REMOVE ANY COVERSHEET OR FORM NOT REQUIRED WITH THE CURRENT FILING BEFORE DELIVERY! Certification Statement: As the adopting Authority of this rule (see 3 V.S.A. § 801 (b) (11) for a definition), I approve the contents of this filing entitled: Vermont Hemp Rules _ (signature) Printed Name and Title: Anson B. Tebbetts, Secretary, Vermont Agency of Agriculture, Food and Markets RECEIVED WV Coversheet Adopting Page doo Economic Impact Analysis Environmental Impact Analysis • Strategy for Maximizing Public Input Scientific Information Statement (if applicable) Incorporated by Reference Statement (if applicable) 00poo Clean text of the rule (Amended text without annotation) Annotated text (Clearly marking changes from previous rule) ICAR Minutes Copy of Comments `---Recetu,cd. -

Scott Sindelar, Ph.D. the Neuropsychology Of
The Neuropsychology of Scott Sindelar, Ph.D. Neuropsychology and Addictions [email protected] Marijuana Chemistry The chemistry of Cannabis is quite complex, and the isolation and extraction of the active ingredients is difficult, even today. Marijuana Chemistry There are 483 chemicals in marijuana, but only 80 of them are unique to the Cannabis plant -- these are called Cannabinoids. Cannabinoids Identified in Marijuana Mohamed M. Radwan, Mahmoud A. ElSohly, et al., researchers at the University of Mississippi, reported in their Apr. 3, 2009 study titled "Biologically Active Cannabinoids from High-Potency Cannabis Sativa,“ the discovery of nine (9) new cannabinoids Cannabinoids Identified in Marijuana Cannabinoid Group Abbreviation Variants 1 9-Tetrahydrocannabinol 9-THC 9 2 8-Tetrahydrocannabinol 8-THC 2 3 Cannabichromene CBC 5 4 Cannabicyclol CBL 3 5 Cannabidiol CBD 7 6 Cannabielsoin CBE 5 7 Cannabigerol CBG 6 8 Cannabinidiol CBND 2 9 Cannabinol CBN 7 10 Cannabitriol CBT 9 11 Miscellaneous types 11 Cannabinoids Identified in Marijuana 80 Cannabinoids discovered Beware of research that states: “THC is the psychoactive ingredient in marijuana” What other pharmaceutical “medication” contains 80 active ingredients? Other constituents of the cannabis plant are: nitrogenous compounds (27 known), amino acids (18), proteins (3), glycoproteins (6), enzymes (2), sugars and related compounds (34), hydrocarbons (50), simple alcohols (7), aldehydes (13), ketones (13), simple acids (21), fatty acids (22), simple esters (12), lactones (1), steroids (11), terpenes (120), non-cannabinoid phenols (25), flavonoids (21), vitamins (1) [Vitamin A], pigments (2), and elements (9). Marijuana smoke contains 50 percent to 70 percent more carcinogenic hydrocarbons than does tobacco smoke and has the potential to cause cancer of the lungs and respiratory tract Mayo Clinic Donald P. -

Vol. 2021, No. 15 I
UTAH STATE BULLETIN OFFICIAL NOTICES OF UTAH STATE GOVERNMENT Filed July 02, 2021, 12:00 a.m. through July 15, 2021, 11:59 p.m. Number 2021-15 August 01, 2021 Nancy L. Lancaster, Managing Editor The Utah State Bulletin (Bulletin) is an official noticing publication of the executive branch of Utah state government. The Office of Administrative Rules, part of the Department of Government Operations, produces the Bulletin under authority of Section 63G-3-402. The Portable Document Format (PDF) version of the Bulletin is the official version. The PDF version of this issue is available at https://rules.utah.gov/. Any discrepancy between the PDF version and other versions will be resolved in favor of the PDF version. Inquiries concerning the substance or applicability of an administrative rule that appears in the Bulletin should be addressed to the contact person for the rule. Questions about the Bulletin or the rulemaking process may be addressed to: Office of Administrative Rules, PO Box 141007, Salt Lake City, Utah 84114-1007, telephone 801-957-7110. Additional rulemaking information and electronic versions of all administrative rule publications are available at https://rules.utah.gov/. The information in this Bulletin is summarized in the Utah State Digest (Digest) of the same volume and issue number. The Digest is available by e-mail subscription or online. Visit https://rules.utah.gov/ for additional information. Office of Administrative Rules, Salt Lake City 84114 Unless otherwise noted, all information presented in this publication is in the public domain and may be reproduced, reprinted, and redistributed as desired. -

Design and Synthesis of Novel Cannabinergic Analogs with Controlled Detoxification
Design and Synthesis of Novel Cannabinergic Analogs with Controlled Detoxification ------------------------------------------------------------------------------ Thesis Presented by Rishi Sharma to The Bouve’ Graduate School of Health Sciences in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Pharmaceutical Sciences with specialization in Medicinal Chemistry and Drug Development NORTHEASTERN UNIVERSITY BOSTON, MASSACHUSETTS April, 2011 Signature page 1 Northeastern University Bouve’ Graduate School of Health Sciences Thesis title: Design and synthesis of novel cannabinergic analogs with controlled detoxification Author: Rishi Sharma Program: Pharmaceutical Sciences with specialization in Medicinal Chemistry and Drug Development Approval for thesis requirement of the Doctor of Philosophy in Pharmaceutical Sciences Thesis Committee (Chairman) ________________________ Date ___________ ________________________ Date ___________ ________________________ Date ___________ ________________________ Date ___________ ________________________ Date ___________ Director of the Graduate School ________________________ Date ___________ Dean ________________________ Date___________ Copy Deposited in Library ________________________ Date___________ Signature page 2 Northeastern University Bouve’ Graduate School of Health Sciences Thesis title: Design and synthesis of novel cannabinergic analogs with controlled detoxification Author: Rishi Sharma Program: Pharmaceutical Sciences with specialization in Medicinal Chemistry -

Cannabis Plant and Cannabis Resin
WHO Expert Committee on Drug Dependence Pre-Review …………….. Cannabis plant and cannabis resin Section 1: Chemistry This report contains the views of an international group of experts, and does not necessarily represent the decisions or the stated policy of the World Health Organization 1 © World Health Organization 2018 All rights reserved. This is an advance copy distributed to the participants of the 40th Expert Committee on Drug Dependence, before it has been formally published by the World Health Organization. The document may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in any form or by any means without the permission of the World Health Organization. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted and dashed lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. The World Health Organization does not warrant that the information contained in this publication is complete and correct and shall not be liable for any damages incurred as a result of its use.