Bacteroidetes Contribute to the Carbon and Nutrient Cycling of Deep Sea
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Polysaccharide-Degrading Marine Bacterium Flammeovirga Sp. MY04 and Its Extracellular Agarase System
中国科技论文在线 http://www.paper.edu.cn J. Ocean Univ. China (Oceanic and Coastal Sea Research) DOI 10.1007/s11802-012-1929-3 ISSN 1672-5182, 2012 11 (3): 375-382 http://www.ouc.edu.cn/xbywb/ E-mail:[email protected] A Polysaccharide-Degrading Marine Bacterium Flammeovirga sp. MY04 and Its Extracellular Agarase System HAN Wenjun1), 2), 3), GU Jingyan1), 3), YAN Qiujie2), LI Jungang2), WU Zhihong3), *, GU Qianqun1), *, and LI Yuezhong3) 1) Key Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, P. R . C hi n a 2) School of Life Science and Biotechnology, Mianyang Normal University, Mianyang 621000, P. R. China 3) State Key Laboratory of Microbial Technology, School of Life Science, Shandong University, Jinan 250100, P. R. China (Received February 2, 2012; revised April 8, 2012; accepted May 29, 2012) © Ocean University of China, Science Press and Springer-Verlag Berlin Heidelberg 2012 Abstract Bacteria of the genus Flammeovirga can digest complex polysaccharides (CPs), but no details have been reported re- garding the CP depolymerases of these bacteria. MY04, an agarolytic marine bacterium isolated from coastal sediments, has been identified as a new member of the genus Flammeovirga. The MY04 strain is able to utilize multiple CPs as a sole carbon source and -1 -1 grows well on agarose, mannan, or xylan. This strain produces high concentrations of extracellular proteins (490 mg L ± 18.2 mg L liquid culture) that exhibit efficient and extensive degradation activities on various polysaccharides, especially agarose. These pro- -1 -1 teins have an activity of 310 U mg ± 9.6 U mg proteins. -

Imperialibacter Roseus Gen. Nov., Sp. Nov., a Novel Bacterium of the Family Flammeovirgaceae Isolated from Permian Groundwater
International Journal of Systematic and Evolutionary Microbiology (2013), 63, 4136–4140 DOI 10.1099/ijs.0.052662-0 Imperialibacter roseus gen. nov., sp. nov., a novel bacterium of the family Flammeovirgaceae isolated from Permian groundwater Hui Wang,1,2,3 Junde Li,1 Tianling Zheng,2 Russell T. Hill3 and Xiaoke Hu1 Correspondence 1Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China Xiaoke Hu 2Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, [email protected] Xiamen University, Xiamen 361005, China 3Institute of Marine and Environmental Technology, University of Maryland Center for Environmental Science, Baltimore, MD 21202, USA A novel bacterial strain, designated P4T, was isolated from Permian groundwater and identified on the basis of its phylogenetic, genotypic, chemotaxonomic and phenotypic characteristics. Cells were aerobic, Gram-stain-negative rods. 16S rRNA gene sequence-based phylogenetic analysis revealed that P4T is affiliated with the family Flammeovirgaceae in the phylum Bacteroidetes, but forms a distinct cluster within this family. The DNA G+C content of strain P4T was 45.2 mol%. The predominant cellular fatty acids were C16 : 1v6c/C16 : 1v7c and iso-C15 : 0. MK-7 was the main respiratory quinone. The polar lipids were phosphatidylethanolamine, phosphatidylglycerol, phosphatidylcholine, unidentified phospholipids, an unidentified aminolipid, unidentified glycoli- pids and unidentified polar lipids. Based on our extensive polyphasic analysis, a novel species in a new genus, Imperialibacter roseus gen. nov., sp. nov., is proposed. The type strain of Imperialibacter roseus is P4T (5CICC 10659T5KCTC 32399T). Bacteria affiliated with the family Flammeovirgaceae of the staining was performed according to the method described phylum Bacteroidetes are widely distributed in various by Gerhardt et al. -

Characterization of a Recombinant Thermostable Arylsulfatase from Deep-Sea Bacterium Flammeovirga Pacifica Chao Gao1†, Min Jin2†, Zhiwei Yi2, and Runying Zeng2,3,4*
J. Microbiol. Biotechnol. (2015), 25(11), 1894–1901 http://dx.doi.org/10.4014/jmb.1504.04028 Research Article Review jmb Characterization of a Recombinant Thermostable Arylsulfatase from Deep-Sea Bacterium Flammeovirga pacifica Chao Gao1†, Min Jin2†, Zhiwei Yi2, and Runying Zeng2,3,4* 1XingHuaLing District Food and Drug Administration, Taiyuan 030000, P.R. China 2State Key Laboratory Breeding Base of Marine Genetic Resource; Third Institute of Oceanography, SOA, Xiamen 361005, P.R. China 3Fujian Collaborative Innovation Center for Exploitation and Utilization of Marine Biological Resources, Xiamen 361005, P.R. China 4South China Sea Bio-Resource Exploitation and Utilization Collaborative Innovation Center, Guangzhou 510000, P.R. China Received: April 13, 2015 Revised: August 4, 2015 A novel sulfatase gene, ary423 (1,536 bp ORF), encoding a protein of 511 amino acids with a Accepted: August 8, 2015 calculated molecular mass of 56 kDa, was identified from Flammeovirga pacifica, which was isolated from deep-sea sediments of west Pacific Ocean. Amino acid sequence analysis revealed that Ary423 possessed a conserved C-X-A-X-R motif, which was recognized as the First published online sulfatase signature. Phylogenetic analysis suggested that Ary423 belonged to arylsulfatases. August 13, 2015 After heterologous expression in Escherichia coli cells, the recombinant Ary423 was purified + *Corresponding author with a Ni affinity column, and was shown to be highly active at a broad range of Phone: +86-592-2195323; temperatures from 30° to 70°C, with maximum activity at 40°C. Furthermore, recombinant Fax: +86-592-2195323; Ary423 retained more than 70% and 40% of its maximum activity after 12 h of incubation at E-mail: [email protected] 50°C and 60°C, respectively, exhibiting good thermostability at high temperatures. -

Carrageenan Catabolism Is Encoded by a Complex Regulon in Marine Heterotrophic Bacteria
ARTICLE DOI: 10.1038/s41467-017-01832-6 OPEN Carrageenan catabolism is encoded by a complex regulon in marine heterotrophic bacteria Elizabeth Ficko-Blean1, Aurélie Préchoux1, François Thomas 1, Tatiana Rochat 2, Robert Larocque1, Yongtao Zhu 3, Mark Stam4, Sabine Génicot1, Murielle Jam1, Alexandra Calteau4, Benjamin Viart4, David Ropartz5, David Pérez-Pascual2, Gaëlle Correc1, Maria Matard-Mann1, Keith A. Stubbs6, Hélène Rogniaux5, Alexandra Jeudy1, Tristan Barbeyron1, Claudine Médigue4, Mirjam Czjzek1, David Vallenet 4, Mark J. McBride3, Eric Duchaud2 & Gurvan Michel 1 1234567890 Macroalgae contribute substantially to primary production in coastal ecosystems. Their biomass, mainly consisting of polysaccharides, is cycled into the environment by marine heterotrophic bacteria using largely uncharacterized mechanisms. Here we describe the complete catabolic pathway for carrageenans, major cell wall polysaccharides of red mac- roalgae, in the marine heterotrophic bacterium Zobellia galactanivorans. Carrageenan cata- bolism relies on a multifaceted carrageenan-induced regulon, including a non-canonical polysaccharide utilization locus (PUL) and genes distal to the PUL, including a susCD-like pair. The carrageenan utilization system is well conserved in marine Bacteroidetes but modified in other phyla of marine heterotrophic bacteria. The core system is completed by additional functions that might be assumed by non-orthologous genes in different species. This complex genetic structure may be the result of multiple evolutionary events including gene duplica- tions and horizontal gene transfers. These results allow for an extension on the definition of bacterial PUL-mediated polysaccharide digestion. 1 Sorbonne Universités, UPMC Univ Paris 06, CNRS, UMR 8227, Integrative Biology of Marine Models, Station Biologique de Roscoff, CS 90074 Roscoff, Bretagne, France. -

Genome-Based Taxonomic Classification Of
ORIGINAL RESEARCH published: 20 December 2016 doi: 10.3389/fmicb.2016.02003 Genome-Based Taxonomic Classification of Bacteroidetes Richard L. Hahnke 1 †, Jan P. Meier-Kolthoff 1 †, Marina García-López 1, Supratim Mukherjee 2, Marcel Huntemann 2, Natalia N. Ivanova 2, Tanja Woyke 2, Nikos C. Kyrpides 2, 3, Hans-Peter Klenk 4 and Markus Göker 1* 1 Department of Microorganisms, Leibniz Institute DSMZ–German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany, 2 Department of Energy Joint Genome Institute (DOE JGI), Walnut Creek, CA, USA, 3 Department of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia, 4 School of Biology, Newcastle University, Newcastle upon Tyne, UK The bacterial phylum Bacteroidetes, characterized by a distinct gliding motility, occurs in a broad variety of ecosystems, habitats, life styles, and physiologies. Accordingly, taxonomic classification of the phylum, based on a limited number of features, proved difficult and controversial in the past, for example, when decisions were based on unresolved phylogenetic trees of the 16S rRNA gene sequence. Here we use a large collection of type-strain genomes from Bacteroidetes and closely related phyla for Edited by: assessing their taxonomy based on the principles of phylogenetic classification and Martin G. Klotz, Queens College, City University of trees inferred from genome-scale data. No significant conflict between 16S rRNA gene New York, USA and whole-genome phylogenetic analysis is found, whereas many but not all of the Reviewed by: involved taxa are supported as monophyletic groups, particularly in the genome-scale Eddie Cytryn, trees. Phenotypic and phylogenomic features support the separation of Balneolaceae Agricultural Research Organization, Israel as new phylum Balneolaeota from Rhodothermaeota and of Saprospiraceae as new John Phillip Bowman, class Saprospiria from Chitinophagia. -
Systematic Bacteriology Second Edition
BERGEY’S MANUAL® OF Systematic Bacteriology Second Edition Volume Four The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes BERGEY’S MANUAL® OF Systematic Bacteriology Second Edition Volume Four The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes Noel R. Krieg, James T. Staley, Daniel R. Brown, Brian P. Hedlund, Bruce J. Paster, Naomi L. Ward, Wolfgang Ludwig and William B. Whitman EDITORS, VOLUME FOUR William B. Whitman DIRECTOR OF THE EDITORIAL OFFICE Aidan C. Parte MANAGING EDITOR EDITORIAL BOARD Michael Goodfellow, Chairman, Peter Kämpfer, Vice Chairman, Jongsik Chun, Paul De Vos, Fred A. Rainey and William B. Whitman WITH CONTRIBUTIONS FROM 129 COLLEAGUES William B. Whitman Bergey’s Manual Trust Department of Microbiology 527 Biological Sciences Building University of Georgia Athens, GA 30602-2605 USA ISBN: 978-0-387-95042-6 e-ISBN: 978-0-387-68572-4 DOI: 10.1007/978-0-387-68572-4 Springer New York Dordrecht Heidelberg London Library of Congress Control Number: 2010936277 © 2010, 1984–1989 Bergey’s Manual Trust Bergey’s Manual is a registered trademark of Bergey’s Manual Trust. All rights reserved. This work may not be translated or copied in whole or in part without the written permission of the publisher (Springer Science+Business Media, LLC, 233 Spring Street, New York, NY 10013, USA), except for brief excerpts in connection with reviews or scholarly analysis. Use in connection with any form of information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed is forbidden. -
Genome Sequencing Reveals the Complex Polysaccharide-Degrading Ability of Novel Deep-Sea Bacterium Flammeovirga Pacifica WPAGA1
ORIGINAL RESEARCH published: 10 April 2017 doi: 10.3389/fmicb.2017.00600 Genome Sequencing Reveals the Complex Polysaccharide-Degrading Ability of Novel Deep-Sea Bacterium Flammeovirga pacifica WPAGA1 Boliang Gao 1, Min Jin 2, Li Li 2, Wu Qu 1 and Runying Zeng 2, 3, 4* 1 School of Life Sciences, Xiamen University, Xiamen, China, 2 State Key Laboratory Breeding Base of Marine Genetic Resource, Third Institute of Oceanography, SOA, Xiamen, China, 3 Fujian Collaborative Innovation Center for Exploitation and Utilization of Marine Biological Resources, Xiamen, China, 4 South China Sea Bio-Resource Exploitation and Utilization Collaborative Innovation Center, Guangzhou, China Flammeovirga pacifica strain WPAGA1 is a Gram-negative, polysaccharide-degrading bacterium isolated from the marine sediment of the West Pacific Ocean. This strain is a cosmopolitan marine bacterium that uses complex polysaccharides as exclusive source of carbon and energy and plays a key role in the marine carbon cycle. Genome sequence Edited by: analysis of strain WPAGA1 revealed that the assembled fine genome contains 6,610,326 Télesphore Sime-Ngando, Centre National de la Recherche bp with 32.89% G+C content, 5036 open reading frames (ORFs) and abundant genomic Scientifique (CNRS), France elements. Amongst these ORFs, 1022 genes encoding carbohydrate enzymes were Reviewed by: found in the F. pacifica WPAGA1 genome. In addition, abundant putative enzymes Claire Geslin, involved in degrading polysaccharide were found. These enzymes include amylase, University of Western Brittany, France Sébastien Monchy, xylosidase, cellulase, alginate lyase, pectate lyase, rhamnogalacturonan lyase, chitinase, Université du Littoral Côte d’Opale, carrageenase, heparinase and fucosidase. To further investigate the use of these France polysaccharides in strain WPAGA1, a schematic of various polysaccharide-degrading *Correspondence: Runying Zeng metabolic pathways were deduced from the genome sequence. -

1989 in Flammeovirga Gen. Nov. As Flammeovirga Aprica Comb. Nov
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, Jan. 1997, p. 220-223 Vol. 47, No. 1 0020-7713/97/$04.00+ 0 Copyright 0 1997, International Union of Microbiological Societies Reclassification of Cytophaga aprica (Lewin 1969) Reichenbach 1989 in Flammeovirga gen. nov. as Flammeovirga aprica comb. nov. and of Cytophaga difluens (ex Stanier 1940; emend. Lewin 1969) Reichenbach 1989 in Persicobacter gen. nov. as Persicobacter difluens comb. nov. YASUYOSHI NAKAGAWA,'" KOEI HAMANA,2 TAKESHI SAKANE,3 AND KAZUHIDE YAMASATO'T Institute of Applied Microbiology$, The University of Tokyo, Bunkyo-ku, Tokyo 113, ' College of Medical Care and Technology, Gunma University, Maebashi 371, and Institute for Fermentation, Osaka, Yodogawa-ku, Osaka 532, Japan Phylogenetically, Cytophugu apricu and Cytophugu difluens occupy independent positions in the flavobacter- bacteroides phylum. Both of these organisms are gram-negative rods that are motile by gliding, chemoor- ganotrophic, and aerobic, degrade several kinds of biomacromolecules, and inhabit marine environments. Their major isoprenoid quinone is menaquinone 7. The G+C content of the DNA of C. upricu is 35 to 37 mol%, and the G+C content of the DNA of C. difluens is 40 to 42 mol%. In addition to constituting an independent phylogenetic lineage, each species has a distinctive cellular polyamine constitution. C. uprica is characterized by possessing cadaverine as its major polyamine, and C. difluens is characterized by possessing spermidine, in contrast to most species of the genera Cytophugu,Fluvobucterium, and Flexibucter and related organisms, which possess homospermidine. Transfer of C. upricu to the genus Flammeovirga gen. nov. as Flammeovirgu upricu comb. nov. and transfer of C. difluens to the genus Persicobucter gen. -

Mooreia Alkaloidigena Gen. Nov., Sp. Nov. and Catalinimonas Alkaloidigena Gen
International Journal of Systematic and Evolutionary Microbiology (2013), 63, 1219–1228 DOI 10.1099/ijs.0.043752-0 Mooreia alkaloidigena gen. nov., sp. nov. and Catalinimonas alkaloidigena gen. nov., sp. nov., alkaloid-producing marine bacteria in the proposed families Mooreiaceae fam. nov. and Catalimonadaceae fam. nov. in the phylum Bacteroidetes Eun Ju Choi, Deanna S. Beatty, Lauren A. Paul, William Fenical and Paul R. Jensen Correspondence Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University Paul R. Jensen of California San Diego, La Jolla, CA 92093-0204, USA [email protected] Bacterial strains CNX-216T and CNU-914T were isolated from marine sediment samples collected from Palmyra Atoll and off Catalina Island, respectively. Both strains were Gram- negative and aerobic and produce deep-orange to pink colonies and alkaloid secondary metabolites. Cells of strain CNX-216T were short, non-motile rods, whereas cells of strain CNU- 914T were short, curved rods with gliding motility. The DNA G+C contents of CNX-216T and CNU-914T were respectively 57.7 and 44.4 mol%. Strains CNX-216T and CNU-914T contained MK-7 as the predominant menaquinone and iso-C15 : 0 and C16 : 1v5c as the major fatty acids. Phylogenetic analyses revealed that both strains belong to the order Cytophagales in the phylum Bacteroidetes. Strain CNX-216T exhibited low 16S rRNA gene sequence identity (87.1 %) to the nearest type strain, Cesiribacter roseus 311T, and formed a well-supported lineage that is outside all currently described families in the order Cytophagales. Strain CNU-914T shared 97.6 % 16S rRNA gene sequence identity with ‘Porifericola rhodea’ N5EA6-3A2B and, together with ‘Tunicatimonas pelagia’ N5DB8-4 and four uncharacterized marine bacteria isolated as part of this study, formed a lineage that is clearly distinguished from other families in the order Cytophagales. -

Characterization of the First Cultured Representative of a Bacteroidetes Clade Specialized on the Scavenging of Cyanobacteria
Characterization of the first cultured representative ofa Bacteroidetes clade specialized on the scavenging of cyanobacteria Wajdi Ben Hania, Manon Joseph, Boyke Bunk, Cathrin Sproer, Hans-Peter Klenk, Marie-Laure Fardeau, Stefan Spring To cite this version: Wajdi Ben Hania, Manon Joseph, Boyke Bunk, Cathrin Sproer, Hans-Peter Klenk, et al.. Character- ization of the first cultured representative of a Bacteroidetes clade specialized on the scavenging of cyanobacteria. Environmental Microbiology, Society for Applied Microbiology and Wiley-Blackwell, 2017, 19 (3), pp.1134 - 1148. 10.1111/1462-2920.13639. hal-01621708 HAL Id: hal-01621708 https://hal-amu.archives-ouvertes.fr/hal-01621708 Submitted on 3 Jul 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Environmental Microbiology (2017) 19(3), 1134–1148 doi:10.1111/1462-2920.13639 Characterization of the first cultured representative of a Bacteroidetes clade specialized on the scavenging of cyanobacteria Wajdi Ben Hania,1 Manon Joseph,1 Boyke Bunk,2 sequences of the MgMjR-022 clade and cyanobacte- Cathrin Sproer,€ 3 Hans-Peter Klenk,4† ria in the suboxic zone of hypersaline mats points to Marie-Laure Fardeau1 and Stefan Spring4* a specific dependence of members of this clade on 1Laboratoire de Microbiologie IRD, MIO, Aix Marseille decaying cyanobacteria. -
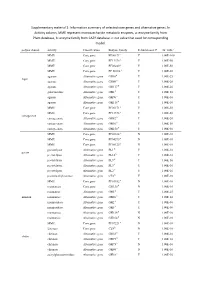
Information Summary of Selected Core Genes and Alternative Genes. in Activity Column, MME Represent Mo
Supplementary material 1: Information summary of selected core genes and alternative genes. In Activity column, MME represent monosaccharide metabolic enzymes, a: enzyme family from Pfam database, b: enzyme family from CAZY database. c: cut value that used for corresponding model. polysaccharide activity Classification Enzyme family Rebuild model? cut_valuec MME Core gene PF00171a Y 1.00E-100 MME Core gene PF13378 a Y 1.00E-50 MME Core gene PF08240 a Y 1.00E-50 MME Core gene PF 00106 a Y 1.00E-50 agarase Alternative gene GH16b Y 1.00E-25 Agar agarase Alternative gene GH86 b Y 1.00E-20 agarase Alternative gene GH117 b Y 1.00E-20 galactosidase Alternative gene GH2 b Y 1.00E-50 agarase Alternative gene GH96 b Y 1.00E-10 agarase Alternative gene GH118 b Y 1.00E-10 MME Core gene PF00171 a Y 1.00E-50 MME Core gene PF13378 a Y 1.00E-50 carrageenan carrageenase Alternative gene GH82 b Y 1.00E-20 carrageenase Alternative gene GH16 b Y 1.00E-50 carrageenase Alternative gene GH150 b Y 1.00E-50 MME Core gene PF02614 a N 1.00E-10 MME Core gene PF04295 a N 1.00E-10 MME Core gene PF08125 a N 1.00E-10 pectatelyase Alternative gene PL1 b Y 1.00E-10 pectin pectatelyase Alternative gene PL10 b Y 1.00E-10 pectatelyase Alternative gene PL9 b Y 1.00E-50 pectatelyase Alternative gene PL3 b Y 1.00E-10 pectatelyase Alternative gene PL2 b Y 1.00E-10 pectinmethylesterase Alternative gene CE8 b Y 1.00E-10 MME Core gene PF01182 a N 1.00E-10 mannanase Core gene GH130 b N 1.00E-10 mannanase Alternative gene GH5 b Y 1.00E-25 mannan mannanase Alternative gene GH26 b -

Genetic Analyses Unravel the Crucial Role of a Horizontally Acquired Alginate Lyase for Brown Algal Biomass Degradation by Z
Genetic analyses unravel the crucial role of a horizontally acquired alginate lyase for brown algal biomass degradation by Z obellia galactanivorans Yongtao Zhu, François Thomas, Robert Larocque, Nan Li, Delphine Duffieux, Lionel Cladière, Florent Souchaud, Gurvan Michel, Mark J. Mcbride To cite this version: Yongtao Zhu, François Thomas, Robert Larocque, Nan Li, Delphine Duffieux, et al.. Genetic analyses unravel the crucial role of a horizontally acquired alginate lyase for brown algal biomass degradation by Z obellia galactanivorans. Environmental Microbiology, Society for Applied Microbiology and Wiley-Blackwell, 2017, 10.1111/1462-2920.13699. hal-01503166 HAL Id: hal-01503166 https://hal.sorbonne-universite.fr/hal-01503166 Submitted on 6 Apr 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Genetic analyses unravel the crucial role of a horizontally acquired alginate lyase for brown algal biomass degradation by Zobellia galactanivorans Yongtao Zhu1#, François Thomas2#, Robert Larocque2, Nan Li3, Delphine Duffieux2, Lionel Cladière2, Florent