A Phylogenomic Profile of Globins Serge N
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Applied Category Theory for Genomics – an Initiative
Applied Category Theory for Genomics { An Initiative Yanying Wu1,2 1Centre for Neural Circuits and Behaviour, University of Oxford, UK 2Department of Physiology, Anatomy and Genetics, University of Oxford, UK 06 Sept, 2020 Abstract The ultimate secret of all lives on earth is hidden in their genomes { a totality of DNA sequences. We currently know the whole genome sequence of many organisms, while our understanding of the genome architecture on a systematic level remains rudimentary. Applied category theory opens a promising way to integrate the humongous amount of heterogeneous informations in genomics, to advance our knowledge regarding genome organization, and to provide us with a deep and holistic view of our own genomes. In this work we explain why applied category theory carries such a hope, and we move on to show how it could actually do so, albeit in baby steps. The manuscript intends to be readable to both mathematicians and biologists, therefore no prior knowledge is required from either side. arXiv:2009.02822v1 [q-bio.GN] 6 Sep 2020 1 Introduction DNA, the genetic material of all living beings on this planet, holds the secret of life. The complete set of DNA sequences in an organism constitutes its genome { the blueprint and instruction manual of that organism, be it a human or fly [1]. Therefore, genomics, which studies the contents and meaning of genomes, has been standing in the central stage of scientific research since its birth. The twentieth century witnessed three milestones of genomics research [1]. It began with the discovery of Mendel's laws of inheritance [2], sparked a climax in the middle with the reveal of DNA double helix structure [3], and ended with the accomplishment of a first draft of complete human genome sequences [4]. -

Brains of Primitive Chordates 439
Brains of Primitive Chordates 439 Brains of Primitive Chordates J C Glover, University of Oslo, Oslo, Norway although providing a more direct link to the evolu- B Fritzsch, Creighton University, Omaha, NE, USA tionary clock, is nevertheless hampered by differing ã 2009 Elsevier Ltd. All rights reserved. rates of evolution, both among species and among genes, and a still largely deficient fossil record. Until recently, it was widely accepted, both on morpholog- ical and molecular grounds, that cephalochordates Introduction and craniates were sister taxons, with urochordates Craniates (which include the sister taxa vertebrata being more distant craniate relatives and with hemi- and hyperotreti, or hagfishes) represent the most chordates being more closely related to echinoderms complex organisms in the chordate phylum, particu- (Figure 1(a)). The molecular data only weakly sup- larly with respect to the organization and function of ported a coherent chordate taxon, however, indicat- the central nervous system. How brain complexity ing that apparent morphological similarities among has arisen during evolution is one of the most chordates are imposed on deep divisions among the fascinating questions facing modern science, and it extant deuterostome taxa. Recent analysis of a sub- speaks directly to the more philosophical question stantially larger number of genes has reversed the of what makes us human. Considerable interest has positions of cephalochordates and urochordates, pro- therefore been directed toward understanding the moting the latter to the most closely related craniate genetic and developmental underpinnings of nervous relatives (Figure 1(b)). system organization in our more ‘primitive’ chordate relatives, in the search for the origins of the vertebrate Comparative Appearance of Brains, brain in a common chordate ancestor. -

Functional Effects Detailed Research Plan
GeCIP Detailed Research Plan Form Background The Genomics England Clinical Interpretation Partnership (GeCIP) brings together researchers, clinicians and trainees from both academia and the NHS to analyse, refine and make new discoveries from the data from the 100,000 Genomes Project. The aims of the partnerships are: 1. To optimise: • clinical data and sample collection • clinical reporting • data validation and interpretation. 2. To improve understanding of the implications of genomic findings and improve the accuracy and reliability of information fed back to patients. To add to knowledge of the genetic basis of disease. 3. To provide a sustainable thriving training environment. The initial wave of GeCIP domains was announced in June 2015 following a first round of applications in January 2015. On the 18th June 2015 we invited the inaugurated GeCIP domains to develop more detailed research plans working closely with Genomics England. These will be used to ensure that the plans are complimentary and add real value across the GeCIP portfolio and address the aims and objectives of the 100,000 Genomes Project. They will be shared with the MRC, Wellcome Trust, NIHR and Cancer Research UK as existing members of the GeCIP Board to give advance warning and manage funding requests to maximise the funds available to each domain. However, formal applications will then be required to be submitted to individual funders. They will allow Genomics England to plan shared core analyses and the required research and computing infrastructure to support the proposed research. They will also form the basis of assessment by the Project’s Access Review Committee, to permit access to data. -

Hmms Representing All Proteins of Known Structure. SCOP Sequence Searches, Alignments and Genome Assignments Julian Gough* and Cyrus Chothia
268–272 Nucleic Acids Research, 2002, Vol. 30, No. 1 © 2002 Oxford University Press SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments Julian Gough* and Cyrus Chothia MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK Received September 28, 2001; Revised and Accepted October 30, 2001 ABSTRACT (currently 59) covering approximately half of the The SUPERFAMILY database contains a library of soluble protein domains. The assignments, super- family breakdown and statistics on them are available hidden Markov models representing all proteins of from the server. The database is currently used by known structure. The database is based on the SCOP this group and others for genome annotation, structural ‘superfamily’ level of protein domain classification genomics, gene prediction and domain-based genomic which groups together the most distantly related studies. proteins which have a common evolutionary ancestor. There is a public server at http://supfam.org which provides three services: sequence searching, INTRODUCTION multiple alignments to sequences of known structure, The SUPERFAMILY database is based on the SCOP (1) and structural assignments to all complete genomes. classification of protein domains. SCOP is a structural domain- Given an amino acid or nucleotide query sequence based heirarchical classification with several levels including the server will return the domain architecture and the ‘superfamily’ level. Proteins grouped together at the super- SCOP classification. The server produces alignments family level are defined as having structural, functional and sequence evidence for a common evolutionary ancestor. It is at of the query sequences with sequences of known this level, as the name suggests, that SUPERFAMILY operates structure, and includes multiple alignments of because it is the level with the most distantly related protein genome and PDB sequences. -

The Origins of Chordate Larvae Donald I Williamson* Marine Biology, University of Liverpool, Liverpool L69 7ZB, United Kingdom
lopmen ve ta e l B Williamson, Cell Dev Biol 2012, 1:1 D io & l l o l g DOI: 10.4172/2168-9296.1000101 e y C Cell & Developmental Biology ISSN: 2168-9296 Research Article Open Access The Origins of Chordate Larvae Donald I Williamson* Marine Biology, University of Liverpool, Liverpool L69 7ZB, United Kingdom Abstract The larval transfer hypothesis states that larvae originated as adults in other taxa and their genomes were transferred by hybridization. It contests the view that larvae and corresponding adults evolved from common ancestors. The present paper reviews the life histories of chordates, and it interprets them in terms of the larval transfer hypothesis. It is the first paper to apply the hypothesis to craniates. I claim that the larvae of tunicates were acquired from adult larvaceans, the larvae of lampreys from adult cephalochordates, the larvae of lungfishes from adult craniate tadpoles, and the larvae of ray-finned fishes from other ray-finned fishes in different families. The occurrence of larvae in some fishes and their absence in others is correlated with reproductive behavior. Adult amphibians evolved from adult fishes, but larval amphibians did not evolve from either adult or larval fishes. I submit that [1] early amphibians had no larvae and that several families of urodeles and one subfamily of anurans have retained direct development, [2] the tadpole larvae of anurans and urodeles were acquired separately from different Mesozoic adult tadpoles, and [3] the post-tadpole larvae of salamanders were acquired from adults of other urodeles. Reptiles, birds and mammals probably evolved from amphibians that never acquired larvae. -

AUTOPHY by Deepika Prasad a Thesis
ANALYZING MARKER GENE DIVERSITY USING AN AUTOMATED PHYLOGENETIC TOOL: AUTOPHY by Deepika Prasad A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Master of Science in Bioinformatics and Computational Biology Spring 2017 © 2017 Deepika Prasad All Rights Reserved ANALYZING MARKER GENE DIVERSITY USING AN AUTOMATED PHYLOGENETIC TOOL: AUTOPHY by Deepika Prasad Approved: __________________________________________________________ Shawn Polson, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Kathleen F.McCoy, Ph.D. Chair of the Department of Computer and Information Sciences Approved: __________________________________________________________ Babatunde A. Ogunnaike, Ph.D. Dean of the College of Engineering Approved: __________________________________________________________ Ann L. Ardis, Ph.D. Senior Vice Provost for Graduate and Professional Education ACKNOWLEDGMENTS I want to express my sincerest gratitude to Professor Shawn Polson for his patience, continuous support, and enthusiasm. I could not have imagined having a better mentor to guide me through the process of my Master’s thesis. I would like to thank my committee members, Professor Eric Wommack and Honzhan Huang for their insightful comments and guidance in the thesis. I would also like to express my gratitude to Barbra Ferrell for asking important questions throughout my thesis research and helping me with the writing process. I would like to thank my friend Sagar Doshi, and lab mates Daniel Nasko, and Prasanna Joglekar, for helping me out with data, presentations, and giving important advice, whenever necessary. Last but not the least, I would like to thank my family, for being my pillars of strength. -

Abstract Introduction
Marine and Freshwater Miscellanea III KEY TRAITS OF AMPHIOXUS SPECIES (CEPHALOCHORDATA) AND THE GOLT1 Daniel Pauly and Elaine Chu Sea Around Us, Institute for the Oceans and Fisheries University of British Columbia, Vancouver, B.C, Canada Email: [email protected] Abstract Major biological traits of amphioxus species (Cephalochordata) are presented with emphasis on the size reached by their 32 valid species in the genera Asymmetron (2 spp.), Branchiostoma (25 spp.), and Epigonichthys (5 spp.) and on related features, i.e., growth parameters and size at first maturity. Overall, these traits combined with features of their respiration, suggest that the cephalochordates conform to the Gill Oxygen Limitation Theory (GOLT), which relates the growth performance of water-breathing ectotherms to the surface area of their respiratory organ(s). Introduction The small fish-like animals know as ‘lancelet‘ or ‘amphioxius’ belong the subphylum Cephalochordata, which is either a sister group, or related to the ancestor of the vertebrate animals (see Garcia-Fernàndez and Benito-Gutierrez 2008). The cephalochordates consist of 3 families (the Asymmetronidae, Epigonichthyidae and Branchiostomidae), with one genus each, Asymmetron (2 spp.), Branchiostoma (24 spp.) and Epigonichthys (6 spp.), as detailed in Table 1 and SeaLifeBase (www.sealifebase.org). This contribution is to assemble some of the basic biological traits of lancelets (Figure 1), notably the maximum size each of their 34 species can reach, which is easily their most important attribute, though it is often ignored (Haldane 1926). Finally, reported lengths at first maturity of cephalochordates were related to the corresponding, population-specific maximum length, to test whether these animals mature as predicted by the Gill- Oxygen Limitation Theory (GOLT; see Pauly 2021a, 2021b). -

Single Cell Chromatin Accessibility of the Developmental
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.17.994954; this version posted March 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Single cell chromatin accessibility of the developmental cephalochordate Dongsheng Chen1, Zhen Huang2,3, Xiangning Ding1,4, Zaoxu Xu1,4, Jixing Zhong1,4, Langchao Liang1,4, Luohao Xu5, Chaochao Cai1,4, Haoyu Wang1,4, Jiaying Qiu1,4, Jiacheng Zhu1,4, Xiaoling Wang1,4, Rong Xiang1,4, Weiying Wu1,4, Peiwen Ding1,4, Feiyue Wang1,4, Qikai Feng1,4, Si Zhou1, Yuting Yuan1, Wendi Wu1, Yanan Yan2,3, Yitao Zhou2,3, Duo Chen2,3, Guang Li6, Shida Zhu1, Fang Chen1,7, Qiujin Zhang2,3, Jihong Wu8,9,10,11 &Xun Xu1 1. BGI-shenzhen, Shenzhen 518083, China 2. Life Science College, Fujian Normal University 3. Key Laboratory of Special Marine Bio-resources Sustainable Utilization of Fujian Province, Fuzhou 350108, P. R. China 4. BGI Education Center, University of Chinese Academy of Sciences, Shenzhen 518083, China 5. Department of Neuroscience and Developmental Biology, University of Vienna 6. State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiangan District, Xiamen, Fujian 361102, China 7. MGI, BGI-Shenzhen, Shenzhen 518083, China 8. Eye Institute, Eye and ENT Hospital, Shanghai Medical College, Fudan University, Shanghai, China 9. Shanghai Key Laboratory of Visual Impairment and Restoration, Science and Technology Commission of Shanghai Municipality, Shanghai, China 10. -

Hemichordate Phylogeny: a Molecular, and Genomic Approach By
Hemichordate Phylogeny: A molecular, and genomic approach by Johanna Taylor Cannon A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama May 4, 2014 Keywords: phylogeny, evolution, Hemichordata, bioinformatics, invertebrates Copyright 2014 by Johanna Taylor Cannon Approved by Kenneth M. Halanych, Chair, Professor of Biological Sciences Jason Bond, Associate Professor of Biological Sciences Leslie Goertzen, Associate Professor of Biological Sciences Scott Santos, Associate Professor of Biological Sciences Abstract The phylogenetic relationships within Hemichordata are significant for understanding the evolution of the deuterostomes. Hemichordates possess several important morphological structures in common with chordates, and they have been fixtures in hypotheses on chordate origins for over 100 years. However, current evidence points to a sister relationship between echinoderms and hemichordates, indicating that these chordate-like features were likely present in the last common ancestor of these groups. Therefore, Hemichordata should be highly informative for studying deuterostome character evolution. Despite their importance for understanding the evolution of chordate-like morphological and developmental features, relationships within hemichordates have been poorly studied. At present, Hemichordata is divided into two classes, the solitary, free-living enteropneust worms, and the colonial, tube- dwelling Pterobranchia. The objective of this dissertation is to elucidate the evolutionary relationships of Hemichordata using multiple datasets. Chapter 1 provides an introduction to Hemichordata and outlines the objectives for the dissertation research. Chapter 2 presents a molecular phylogeny of hemichordates based on nuclear ribosomal 18S rDNA and two mitochondrial genes. In this chapter, we suggest that deep-sea family Saxipendiidae is nested within Harrimaniidae, and Torquaratoridae is affiliated with Ptychoderidae. -

Molecular Investigation of the Cnidarian-Dinoflagellate Symbiosis
AN ABSTRACT OF THE DISSERTATION OF Laura Lynn Hauck for the degree of Doctor of Philosophy in Zoology presented on March 20, 2007. Title: Molecular Investigation of the Cnidarian-dinoflagellate Symbiosis and the Identification of Genes Differentially Expressed during Bleaching in the Coral Montipora capitata. Abstract approved: _________________________________________ Virginia M. Weis Cnidarians, such as anemones and corals, engage in an intracellular symbiosis with photosynthetic dinoflagellates. Corals form both the trophic and structural foundation of reef ecosystems. Despite their environmental importance, little is known about the molecular basis of this symbiosis. In this dissertation we explored the cnidarian- dinoflagellate symbiosis from two perspectives: 1) by examining the gene, CnidEF, which was thought to be induced during symbiosis, and 2) by profiling the gene expression patterns of a coral during the break down of symbiosis, which is called bleaching. The first chapter characterizes a novel EF-hand cDNA, CnidEF, from the anemone Anthopleura elegantissima. CnidEF was found to contain two EF-hand motifs. A combination of bioinformatic and molecular phylogenetic analyses were used to compare CnidEF to EF-hand proteins in other organisms. The closest homologues identified from these analyses were a luciferin binding protein involved in the bioluminescence of the anthozoan Renilla reniformis, and a sarcoplasmic calcium- binding protein involved in fluorescence of the annelid worm Nereis diversicolor. Northern blot analysis refuted link of the regulation of this gene to the symbiotic state. The second and third chapters of this dissertation are devoted to identifying those genes that are induced or repressed as a function of coral bleaching. In the first of these two studies we created a 2,304 feature custom DNA microarray platform from a cDNA subtracted library made from experimentally bleached Montipora capitata, which was then used for high-throughput screening of the subtracted library. -

An EST Screen from the Annelid Pomatoceros Lamarckii Reveals
BMC Evolutionary Biology BioMed Central Research article Open Access An EST screen from the annelid Pomatoceros lamarckii reveals patterns of gene loss and gain in animals Tokiharu Takahashi*1,4, Carmel McDougall2,4,6, Jolyon Troscianko3, Wei- Chung Chen4, Ahamarshan Jayaraman-Nagarajan5, Sebastian M Shimeld4 and David EK Ferrier*2,4 Address: 1Faculty of Life Sciences, University of Manchester, Oxford Road, Manchester, UK, 2The Scottish Oceans Institute, University of St. Andrews, St. Andrews, Fife, UK, 3Centre for Ornithology, School of Biosciences, University of Birmingham, Edgbaston, Birmingham, UK, 4Department of Zoology, University of Oxford, South Parks Road, Oxford, UK, 5Department of Biochemistry, University of Oxford, South Parks Road, Oxford, UK and 6School of Biological Sciences, University of Queensland, St Lucia, Queensland, Australia Email: Tokiharu Takahashi* - [email protected]; Carmel McDougall - [email protected]; Jolyon Troscianko - [email protected]; Wei-Chung Chen - [email protected]; Ahamarshan Jayaraman- Nagarajan - [email protected]; Sebastian M Shimeld - [email protected]; David EK Ferrier* - dekf@st- andrews.ac.uk * Corresponding authors Published: 25 September 2009 Received: 1 April 2009 Accepted: 25 September 2009 BMC Evolutionary Biology 2009, 9:240 doi:10.1186/1471-2148-9-240 This article is available from: http://www.biomedcentral.com/1471-2148/9/240 © 2009 Takahashi et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -
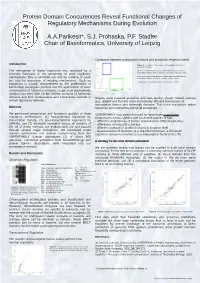
Protein Domain Coocurences Reveal Functional Changes of Regulatory Mechanisms During Evolution
Protein Domain Coocurences Reveal Functional Changes of Regulatory Mechanisms During Evolution A.A.Parikesit*, S.J. Prohaska, P.F. Stadler Chair of Bioinformatics, University of Leipzig Correlation between transcription factors and chromatin related proteins Introduction Figure 3 : Correlation of the number of transcription factors and chromatin domains. The emergence of higher organisms was facilitated by a Blue box. Species with few chromatin-related domains but many dramatic increases in the complexity of gene regulatory transcription factors: human, kangaroo rat, mouse, opossum, fugu. Green box: species with many chromatin-related domains but few mechanisms. This is achieved not only by addition of novel transcription factors: seq squirt, medaka, dolphin, yeast but also by expansion of existing mechanisms. Such an There are no organisms (known) that have a lot of both. expansion is usually characterized by the proliferation of functionally paralogous proteins and the appearance of novel combinations of functional domains. Large scale phylogenetic analysis can shed light on the relative amounts of functional domains and their combinations and interactions involved in Results show massive problems with data quality: closely related species certain regulatory networks. (e.g, dolphin and human) show dramatically different distributions of transcription factors and chromatin domains. This is not reasonable within Methods mammals and contradicts biological knowledge. We performed comparative and functional analysis of three SUPERFAMILY thus cannot be used for large-scale quantitative regulatory mechanisms: (1) transcriptional regulation by comparisons across species due to several sources of bias: transcription factors, (2) post-transcriptional regulation by - different completeness of protein annotation for different genomes miRNAs, and (3) chromatin regulation across all domains of - differences in transcript coverage life.