Pakistan Armed Forces Medical Journal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Challenges and Opportunities of Multilingual Library Websites: Evidence from a Developing Country
International Journal of Business and Management Invention ISSN (Online): 2319 – 8028, ISSN (Print): 2319 – 801X www.ijbmi.org Volume 1 Issue 1 ‖‖ December. 2012 ‖‖ PP.01-15 Challenges and Opportunities of Multilingual Library Websites: Evidence from a Developing Country Amir Manzoor (Management Sciences Department, Bahria University, Karachi, Pakistan) ABSTRACT: This study investigates the language barriers on library websites, analyzingdata collected from 133 library websites in Pakistan. Despite a significant increase in the number of library websites in Pakistan, the development of multilingual library websites is still in its infancy stages and doesn’t cater to the needs of diverse population of its users. The study provides first-hand findings on the multilinguality of library websites in Pakistan with a focus on challenges and opportunities arising from such websites. Implications of research along with future research directions are also discussed. Keywords––Language Barriers, Library Websites, Multilingual Websites, Machine Translation. I. INTRODUCTION Language barriers are natural in human communication. In cross border communication, these barriers not only include dominance of a particular language (e.g. English) but also the increasingly multilingual the Internet [(David Crystal 2001); (Becker 2007)]. Since beginning of the last decade, the multilinguality on the Internet has sharply increased with online content available in hundreds of languages [(Crystal 2006); (Kelsey 2011)]. This increased multilinguality has created new challenges for language barriers. In order to facilitate the multilingual users to effectively navigate, search, filter, and retrieve the multilingual content from the library websites, various stakeholders need to do collective efforts [(Dubois 1979);(Yunker 2002); (Tixier 2005); (Becker 2007); (Diekema 2012)]. -

NUSTNEWS Volume IV / Issue IX
National University of Sciences & Technology September 2013 MONTHLY NUSTNEWS Volume IV / Issue IX 5th World Engineering Congress at NUST Page 03 First Aid Workshop Orientation Sessions Intra-NUST for Freshmen Badminton Championship Page 13 Page 20 Page 23 Soft copy can be downloaded from NUST website: www.nust.edu.pk/downloads NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS NUSTNEWS With ordinary talent “and extraordinary perseverance, all“ things are attainable. Thomas Fowell Buxton CONTENTS 03-13 HIGHLIGHTS 14-22 UPDATES FROM SCHOOLS 23-25 CO-CURRICULARS CORNER 26-27 ACHIEVEMENTS Editorial Team Editor: Maryam Khalid Assistant Editor: Faheem Khaliqdad Graphics & Layout: Kareem Muhammad Photography: Ghulam Rasool NUSTNEWS is a monthly publication, produced by Student Affairs Directorate, covering various events across the entire Student Reporters: University. It will be appreciated if the focal Zaid Bin Khamis Butt, Anum Yousaf Khan, persons send reports right after the events so Taimoor Ahmad as to give them timely coverage. NUSTNEWS NUSTNEWS / SEPTEMBER 2013 3 01 HIGHLIGHTS Fifth World Engineering Congress-2013 at NUST Against the backdrop of a myriad of challenges and the alarm- Federal Minister for Science and Technology, Zahid Hamid also ing scenario unfolding as a consequence, the engineering pro- dilated upon the greater significance engineering and technol- fession holds the key to reverse the crises that may drag the ogy has assumed as a result of population explosion especially world into a chaotic situation forbidding the human life to exist in the developing countries. He was of the view that the de- at ease. As the world population continues to multiply at a pace liberations made during the Congress would contribute to the much faster than ever, engineering remedies to the ever-grow- overall development of the engineering sector, which serves as ing fundamental problems like nutrition, health and education the backbone of any economy. -

Effectiveness of Cardiac Rehabilitation on Health-Related
ORIGINAL ARTICLE Effectiveness of Cardiac Rehabilitation on Health-related Quality of Life in Patients with Myocardial Infarction in Pakistan Zia Ul-Haq1,4, Daud Khan1, Aliya Hisam2, Yasar Mehmood Yousafzai1, Shazia Hafeez3, Fatima Zulfiqar3, Adnan Mahmood Gul3, Mohammad Hafizullah1 and Jill Pell4 1Institute of Public Health & Social Sciences, Khyber Medical University, Peshawar, Pakistan 2Department of Community Medicine, Army Medical College, National University of Medical Sciences, Rawalpindi, Pakistan 3Department of Preventive Cardiology, Lady Reading Hospital, Peshawar, Pakistan 4Institutes of Health & Wellbeing, University of Glasgow, UK ABSTRACT Objective: To find out the effectiveness of cardiac rehabilitation in patients with myocardial infarction in Pakistan. Study Design: Randomised controlled trial. Place and Duration of Study: Cardiac Rehabilitation Unit, Lady Reading Hospital, Peshawar, Pakistan, from July to December 2016. Methodology: Patients suffering first myocardial infarction (MI) were randomly allocated to usual care or cardiac rehabilitation in a 1:1 ratio. Cardiac rehabilitation comprised two phases: 1-2 weeks during hospital stay followed by 6-7 weeks outpatient structured exercise programme. Two generic health related quality of life (HRQoL) outcomes (General Health Questionnaire (GHQ) and Self-Rated Health (SRH)) and one post-MI specific tool (MacNew QLMI) were measured at baseline and at 8 weeks follow-up among both groups. Lower SRH and GHQ scores and higher MacNew QLMI scores indicate better health status. Data were analysed using STATA 14. Results: Out of 206 participants, 195 (94.6%) were analysed at the end of trial. The mean age was 53 +8.3 years. In the cardiac rehabilitation group, the mean SRH score changed from 3.97 +0.9 at baseline to 2.36 +0.8 at follow-up (p<0.001). -

Pattern of Coagulation Parameters In
Open Access Original Article Coagulation Parameters in Patients With COVID-19 Pak Armed Forces Med J 2020; 70 COVID-19 (1): S285-91 PATTERN OF COAGULATION PARAMETERS IN PATIENTS WITH COVID-19 -A SINGLE CENTRE BASED STUDY Nimra Anwar, Fahim Akhtar, Sunila Tashfeen, Kehkashan Hassan, Hafeez Ud Din, Javaid Usman Army Medical College/National University of Medical Sciences (NUMS) Rawalpindi Pakistan ABSTRACT Objective: To determine prognostic significance of coagulation parameters in patients with COVID-19. Study Design: A prospective comparative study. Place and Duration of Study: Department of Haematology, Army Medical College, Pak Emirates Military Hospital, Rawalpindi, from Apr to May 2020. Methodology: A total of 248 patients diagnosed with COVID-19 of all ages irrespective of gender were enrolled. Their coagulation parameters were assessed and comparisons were made between patients with mild/moderate (non-critical) disease against those with severe/critically ill (critical).Performa was designed and data was analyzed using SPSS 26. Results: Patients in the critical group revealed constantly elevated levels of Domain-dimer (D-dimer, ng/ml - 73.7% vs. 50.5%, 89.5% vs. 39%, 78.9 vs. 41.9%, 77.8% vs. 42%), increased activated partial thromboplastin time (APTT - 34.68 vs. 32.17 sec, 38.84 vs. 32.40 sec, 37.58 vs. 32.50 sec , 37.94 vs. 32.61 sec) and prothrombin time (PT - 14.26 vs. 14.20 sec, 14.79 vs. 14.08 sec, 14.68 vs. 14.10 sec, 15 vs. 14.25 sec) compared to noncritical group (p<0.05). Moreover, higher fibrinogen levels were associated with severe disease (296.32 vs257.92 mg/dl, 280.53 vs. -

National University of Medical Sciences (NUMS) Entry Test For
National University of Medical Sciences (NUMS) Entry Test for MBBS/BDS Admissions 2017 Program College / Institute Basic Qualification/ Eligibility MBBS Constituent: - Minimum 60% aggregate marks each in Matric, F.Sc (Pre-Medical) OR Army Medical College, (AM College) Equivalent. Rawalpindi Candidates awaiting result of Intermediate (Pre-Medical) annual Examination Affiliated Institutions: - 2017 can apply on the basis of F.Sc Part-I. However, confirmation of CMH Lahore Medical College & admissions is subject to provision of F.Sc Certificate with detailed marks Institute of Dentistry, Lahore Certificate. CMH Multan Institute of Medical Candidates of A level (science group) or equivalent qualification, appearing in Sciences, (CIMS) Multan final examinations, can apply on the basis of O level / equivalent qualifications Quetta Institute of Medical Sciences, and equivalence certificate from Inter Board Committee of Chairmen (IBCC) (QIMS) Quetta office but confirmation of admissions will be on provision of equivalence WAH Medical College, Wah Cantt certificate for A Level /equivalent qualification. CMH Institute of Medical Sciences Foreign Students can apply on basis of SAT-II Examination with minimum score (CIMS) Bahawalpur of 550 marks in each of the three science subjects of which two have to be HITECH Institute of Medical Biology and Chemistry OR MCAT with minimum aggregate score of 24. Sciences, Taxila All Pakistani Nationals including Azad Jammu & Kashmir and Gilgit Baltistan Karachi Institute of Medical Sciences, (KIMS) -

Batch-13 Candidates Waiting for Exam
Batch-13 Candidates Waiting For Exam Note: The following Candidates are advised to consciously keep on checking their email and sms because intimation about Exam schedule would be made to each candidate by Virtual University through sms and email, subject to the availability of Examination Center under GOP SOP in prevailing Pandemic scenario. Hence, no need to contact VU or NITB in this regard. S.No App_ID Off_Sr Name Course_For Department 1 64988 17254 Zeeshan Ullah LDC/UDC 301 Spares Depot EME Golra Morh Rawalpindi 2 64923 17262 Muhammad Harris LDC/UDC 301 Spares Depot EME Golra Morh Rawalpindi 3 64945 17261 Muhammad Tahir LDC/UDC 301 Spares Depot EME Golra Morh Rawalpindi 4 62575 16485 Aamir Javed LDC/UDC 304 Spares Depot EME, Khanewal 5 63798 16471 Jaffar Hussain Assistant 304 Spares Depot EME, Khanewal 6 64383 17023 Asim Ismail LDC/UDC 501 Central Workshop EME Rawalpindi 7 64685 17024 Shahrukh LDC/UDC 501 Central Workshop EME Rawalpindi 8 64464 17431 Muhammad Shahid Khan LDC/UDC 502 Central work shop EME 9 29560 17891 Asif Ehsan LDC/UDC 602 Regional work shop EME 10 64540 17036 Imran Qamar LDC/UDC 602 Regional Workshop EME 11 29771 17889 Muhammad Shahroz LDC/UDC 602 Regional Workshop EME 12 29772 17890 Muhammad Faizan LDC/UDC 602 Regional Workshop EME 13 63662 16523 Saleh Muhammad Assistant 770 LAD EME Junior Leader Academy Shinikiari Muhammad Naeem 14 65792 18156 Ahmed Assistant Academy of Educational Planning and Management 15 64378 18100 Malik Muhammad Bilal LDC/UDC Administrative and Management office GHQ Rawalpind 16 64468 16815 -

College Inspection Report
PAKISTAN MEDICAL COMMISSION LAST RECOGNIZED INSPECTION GRADES OF PRIVATE MEDICAL COLLEGES Sr. Date of Previous Name of Institute City Grade No. Inspection 1. Abbottabad International Medical College Abbottabad. 17-12-2019 A 2. Abwa Medical College Faisalabad 20-11-2018 B 3. Aga Khan University Medical College Karachi. 05-08-2019 A+ 4. Akhtar Saeed Medical & Dental College Lahore. 26-08-2019 A 5. Al Aleem Medical College Lahore 29-08-2019 B 6. Al-Nafees Medical College Islamabad. 30-12-2015 C 7. Al-Tibri Medical College Karachi. 08-11-2013 B 8. Amna Inayat Medical College Lahore. 28-09-2017 C 9. Avicenna Medical College Lahore. 28-08-2019 A 10. Aziz Fatimah Medical & Dental College Faisalabad. 21-03-2018 C 11. Azra Naheed Medical College Lahore. 30-04-2015 B 12. Bahria University Medical College Karachi. 07-08-2019 B 13. Bakhtawar Amin Medical & Dental College Multan 20-02-2016 A 14. Baqai Medical College Karachi. 19-12-2018 F 15. Central Parks Medical College Lahore. 10-02-2015 C 16. CMH Institute of Medical Sciences Bahawalpur Bahawalpur 22-08-2019 C 17. CMH Kharian Medical College Kharian Cantt. 31-07-2019 B 18. CMH Lahore Medical College Lahore Cantt. 30-08-2019 A+ 19. CMH Multan Institute of Medical Sciences CIMS Multan Cantt 20-08-2019 A 20. Continental Medical College Lahore. 16-10-2018 C GRADING CRITERIA: 92.5% or above = A+, 85% or above = A, 77.5% or above = B, 70% or above = C, 69.9% or lower = F Sr. Date of Previous Name of Institute City Grade No. -

PRESIDENTS MESSAGE Parvez I
Raana Akbar, M.D., Editor EXECUTIVE COUNCIL Officers 4121 Fairview Ave., Downers Grove, IL 60515; Tele: 708/968-8585 or Fax #: 708/968-8677 President Mushtaq A. Khan, M.D. VOLUME I NUMBER 2 Office: 1815) 7-26-6611 SEPTEMBER/OCTOBER 1991 Home: (708) 323-3564 President-Elect PRESIDENTS MESSAGE Parvez I. Shah, M.D. Office: (301) 490-0500 Dear Members: Assalam Alaikum! one. The APPNA "12th International Home: (301) 384-9203 Secretary Health Conference" will take place M. Khalid Riaz, M.D. This summer again has seen the on December 17, 18, 19, 1991 in Office: 1708) 931-4200 Home: 1708) 382-4667 celebration of Pakistan Independence Day Rawalpindi/Islamabad, arrangements Treasurer in various cities of North America. The are progressing well. In the previous M. M. Hague, M.D. Office: (708) 323-5800 enthusiasm for this occasion has been Newsletter registration form and a call Home: (708) 986-8212 contagious. The festivities have included for abstracts were mailed to the Immediate Past-President Arif M. Muslim, M.D. parades, rallies and various functions in membership. If you have not responded Office: (914) 562-0740 Home: (914) 565-3266 which many dignitaries took part. yet, please take a moment to make Alumni Association Concerted community efforts of several arrangements as soon as possible. Presidents years bore fruits in Chicago where a portion Azizur R. Arain, M.D. (Dow) of popular Devon Street was renamed The APPNA Central Office is equipped Office: (708) 971-3323 Bushra QuresM; M.D. Mohammad Ali Jinnah Way. with a fax machine, APPNA membership (Fatima )innah) Congratulations! Office: (416) 461-9471 is not fully utilizing this amazing Shahid I. -
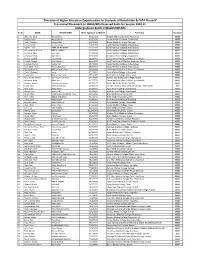
Balochistan & FATA Phase-II" Provisional Placement on MBBS/BDS Reserved Seats for Session 2020-21 Undergraduate Batch-V (BALOCHISTAN)
"Provision of Higher Education Opportunities for Students of Balochistan & FATA Phase-II" Provisional Placement on MBBS/BDS Reserved Seats for Session 2020-21 Undergraduate Batch-V (BALOCHISTAN) Sr. No NAME FATHER NAME Merit Aggregate w/MDCAT Placement Discipline 1 Wasiq Ali Tariq Tariq Bashir 93.627273 Khyber Medical College, Peshawar. MBBS 2 Rimsha Khan Nasrullah Jan 93.613636 Army Medical College, Rawalpindi. MBBS 3 Abdul Qayyum Naeem khan 93.536364 Khyber Medical College, Peshawar. MBBS 4 Asmat Ullah shams ul haq 92.845455 Ayub Medical College, Abbottabad. MBBS 5 Najeeb Ullah AMIR MUHAMMAD 92.272727 Ayub Medical College, Abbottabad. MBBS 6 Muhammad Ibrahim ABDUL JABBAR 91.650000 Ayub Medical College, Abbottabad. MBBS 7 Mohabat Khan Sado khan 91.013636 Ayub Medical College, Abbottabad. MBBS 8 Arfa Ayaz Khan Ayaz Khan 90.581818 Army Medical College, Rawalpindi. MBBS 9 Ayesha Baloch Ejaz Ahmed 90.145455 Gujranwala Medical College, Gujranwala. MBBS 10 Farooq Ahmed Altaf Hussain 89.022727 KUST Institute of Medical Sciences, Kohat. MBBS 11 Sundeep Kumar Vadoo Mal 88.740909 Ayub Medical College, Abbottabad. MBBS 12 Sana Ullah Khan Raz Muhammad 88.254545 Ayub Medical College, Abbottabad. MBBS 13 Muhammad Ali Muhammad Safdar 88.181818 Gujranwala Medical College, Gujranwala. MBBS 14 Abdul Waheed abdul 87.159091 Ayub Medical College, Abbottabad. MBBS 15 Tariq Iqbal Muhammad iqbal 85.518182 Ayub Medical College, Abbottabad. MBBS 16 Syed Haider Ali Shah Syed Asghar Ali Shah 84.440909 Nawaz Sharif Medical College, Gujrat. MBBS 17 Muheem Khan Mian dad 84.277273 Gujranwala Medical College, Gujranwala. MBBS 18 Alveena Azeem Muhammad Azeem 83.704545 Bolan Medical College, Quetta. -

Inflammation of Interstitial Connective Tissue Space Induced By
Open Access Original Article Interstitial Connective Tissue Space Pak Armed Forces Med J 2021; 71 (3): 1066-70 INFLAMMATION OF INTERSTITIAL CONNECTIVE TISSUE SPACE INDUCED BY LEAD ACETATE AND AMELIORATIVE AFTERMATH OF FICUSCARICAON RAT TESTIS Ayesha Asad, Shabnam Hamid*, Afnan Gul**, Noreen Anwar, Dujanah Bhatti***, Naureen Waseem**** Quetta Institute of Medical Sciences, Quetta Pakistan, *Army Medical College/National Institute of Medical Sciences (NUMS) Rawalpindi Pakistan, **Shalamar Medical College Lahore Pakistan, ***Holy Family Hospital, Rawalpindi Pakistan, ****Islam Medical College, Sialkot Pakistan ABSTRACT Objective: To observe inflammation of interstitial connective tissue space caused by Lead acetate in rat testis and ameliorative after math caused by Ficuscarica. Study Design: Laboratory based experimental study. Place and Duration of Study: Anatomy Department, Army Medical College Rawalpindi together with NIH (National Institute of Health) Islamabad, from Mar to Nov 2017. Methodology: Sprague Dawley male rats, 30 in quantity were chosen and 10 animals each partited into 3 groups. Treatments were given for 8 weeks, once daily. Group A was control group. Group B was treated with dosage of 30 mg/kg of Lead acetate. Group C was given dosage of 30 mg/kg of Lead acetate as well as 80 mg/kg of Ficuscarica. Twenty four hours after the concluding dose, animals were vivisected. For histological study, testis were fixed and stained with Haematoxylin and eosin. Interstitial connective tissue space thickness was morphometrically and assessed by SPSS version 22. A p-value <0.05 was considered significant in results. Results: Interstitial space thickness was significantly increased due to inflammation (>3 times normal) in group B in compa- rison to groups A and C. -

Updated-TCF-Scholarship-Policy-For
Appendix A TIERS MEDICAL ENGG COMPUTERS BUSINESS ARTS & HUMANITIES OTHERS GIKI, NUST, GIKI, NUCES, ICAP, ACCA, ACTUARIAL NUCES, UET, NUST, TIER 1 DOW, AKUH, KE LUMS IVS, NCA, LUMS, HU SCIENCES, CFA, CSS, PIEAS, PU, NED, COMSATS, ARMED FORCES LUMS LUMS Mehran QOU, AIFD, TIP, PU, ZIAUDDIN, University, NED, BAHRIA, NBS,LSE, PU, FITFD, LSFD, KU, TIER 2 BAQAI, SMC, Dawood, Sir KU BAHRIA, KU Sindh University AMC, LUHS Syed Jamshoro JPMC, LNMH, PAF KIET, IQRA, NED, Federal Urdu TIER 3 IEEE, DHA Suffa IQRA, PAF KIET KMDC, UHS-LHR DHA Suffa University Appendix B Jamshoro Punjab University TIERS KU PU QU FC GC NED University College of Sargodha Visual Studies, Pharmacy, BS Physics, Actuarial Engg, BBA BS in Associate IR, Strategic BBA, B.Sc in Sciences, IR, (IBA), Business, Political B.E TIER Degree Studies, Pharmacy, Mass Comm, IBIT, Law, Economic Science, (Electri 1 Programmes Political Mechanical English, Journalis s, BSC, BS English cal & All , B.Ed Science, and Microbiology m, in Literature, Mecha (Hons), BFA Economics, Electrical , Psychology, Political Biological History nical) (Hons) Physics Engineerin BBA, UBIT, Science Sciences g Public Adm, Economics, Maths BA/BS in Education South Asian , BS in University – English, Anthropolo Central Mass Compu Islamic gy, Pakistan TIER Punjab Communi ter, IT, Studies, CS, Studies, 2 cation, Bio- Botany History, (All Chemistry tech BBA, BPA Programmes , Physics, ) Environm ental Sciences Sociology, Political Geograph Science, y, History, Sociology, Political Education, Science, TIER Geography, Psycholog -

Pakistan Armed Forces Medical Journal Contents
PAKISTAN ARMED FORCES MEDICAL JOURNAL Vol-70, No. 4, August 2020 Recognized by PMDC & HEC CONTENTS EDITORIAL Differentiating Between Mad And Bad: Forensic Psychiatry Services May Be Need of The Hour 878 Editorial Advisory Board Usama Bin Zubair Chairman ORIGINAL ARTICLES Frequency of Headache And Improvement With The Treatment Among The Patients of Epilepsy 880 Lt Gen Nigar Johar, HI (M) (Surg Gen/DGMS (IS) Fatima Naseem Mailk, Usama Bin Zubair, Khurram Haq Nawaz, Saeed Arif, Asif Hashmat, Wasim Alamgir Members Lipid-Lowering Efficacy of 20Mg Simvastatin Versus 5 Mg Rosuvastatin in Patients of Type 2 Diabetes Prof Irfan Ali Sheikh Hafiz Muhammad Yasir Rehman, Abdul Latif Khattak, Taimoor Ashraf Khan, Abdul Moueed Tariq, 885 Shahzeb Ahmed Satti, Rafi Ud Din Prof Imran Fazal Guillain-Barré Syndrome; The Seasonal Trends in A Cohort of Pakistani Population From Southern Dr Salman Ashraf Punjab 890 Dr Qamar Ul Haq Noor Nadeem Ahmad, Zaheer Ahmed Gill, Saeed Bin Ayaz, Noreen Akhtar, Aamir Waheed Butt, Waseem Iqbal Dr Zaheer Akhtar Maternal And Perinatal Outcome in Pregnancy Induced Hypertensive Mothers in Combined Military Hospital, Sialkot 896 Dr Sibtain Rafique Jehan Ara Rafiq Baig, Muhammad Musarrat Jamal Patient Safety; Interventions To Reduce Hospital Errors Editorial Committee 902 Sana Abbas, Amjad Akram, Syed Hamid Ali Shah, Rashid Iqbal, Beenish Abbas, Uzma Urooj Chief Editor Experience of Endoscopic Repair of Cerebrospinal Fluid Rhinorrhea 910 Prof Sayed Nusrat Raza Anwar ul Haq, Muhammad Khan, Asif Alam Gul, Adnan Asghar Editor Evaluation