Summary of the AOP
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rotenone-Induced Inner Retinal Degeneration Via Presynaptic
www.nature.com/scientificreports OPEN Rotenone-induced inner retinal degeneration via presynaptic activation of voltage-dependent sodium and L-type calcium channels in rats Masaaki Sasaoka, Takashi Ota & Masaaki Kageyama* Rotenone, a mitochondrial complex I inhibitor, causes retinal degeneration via unknown mechanisms. To elucidate the molecular mechanisms of its action, we further characterized a rat model of rotenone- induced retinal degeneration. Intravitreal injection of rotenone (2 nmol/eye) damaged mainly the inner retinal layers, including cell loss in the ganglion cell and inner nuclear layers, which were very similar to those induced by 10 nmol/eye N-methyl-D-aspartate (NMDA). These morphological changes were accompanied by the reduced b-wave amplitude of electroretinogram, and increased immunostaining of 2,4-dinitrophenyl, an oxidative stress marker. Rotenone also downregulated expression of neuroflament light-chain gene (Nf) as a retinal ganglion cell (RGC) marker. This efect was prevented by simultaneous injection of rotenone with antioxidants or NMDA receptor antagonists. More importantly, voltage-dependent sodium and L-type calcium channel blockers and intracellular calcium signaling modulators remarkably suppressed rotenone-induced Nf downregulation, whereas none of these agents modifed NMDA-induced Nf downregulation. These results suggest that rotenone-induced inner retinal degeneration stems from indirect postsynaptic NMDA stimulation that is triggered by oxidative stress-mediated presynaptic intracellular calcium signaling via activation of voltage-dependent sodium and L-type calcium channels. Rotenone is a naturally occurring and broad-spectrum pesticide that inhibits the activity of NADH dehydroge- nase in the mitochondrial respiratory chain complex I1. Because of this unique biological activity, rotenone has been used as a versatile tool to study involvement of mitochondrial functions and oxidative stress in neuronal cell death. -

A Radical Approach to Stroke Therapy
Commentary A radical approach to stroke therapy James McCulloch* and Deborah Dewar Wellcome Surgical Institute and Hugh Fraser Neuroscience Laboratories, University of Glasgow, Glasgow G61 1QH, United Kingdom troke is a major cause of death and readily crosses the blood–brain barrier, (mitogen-activated protein kinases͞ Sdisability throughout the developed to augment endogenous brain ascorbic ERK1͞2). During ischemia, ERK1͞2 are world. Cerebrovascular disease ranks acid levels by up to 2 mM. However, in dephosphorylated, and there is a signif- third after cancer and heart disease as a the brain, ascorbic acid levels are heter- icant increase in ERK1͞2 phosphoryla- cause of death in the European Union ogeneous and highly compartmental- tion during reperfusion after forebrain and the U.S. The economic and social ized. In the rat, normal neuronal, glial, ischemia. Neurons and oligodendrocytes burdens of stroke are not consequences and cerebrospinal fluid levels of ascorbic at the margin of a focal ischemic lesion of mortality; they are imposed by the acid are Ϸ10 mM, 1 mM, and 0.5 mM, display increased MEK1͞2, indicating large majority of stroke patients who respectively (3). The antioxidant effects that this signaling pathway is activated survive but are physically and mentally of ascorbic acid at all of these sites within after ischemia and reperfusion in vivo disabled by stroke-induced brain dam- the brain could contribute to the efficacy (8). If MEK1͞2 is inhibited by the novel age. In the U.S., less than 2% of stroke of this agent in ischemia. Brain ascorbic agent, U0126, the extent of brain damage patients benefit from access to early acid levels are highly dynamic (although is reduced after either forebrain or focal thrombolysis, which removes the primary under rigorous homeostatic control), ischemia (2). -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Evaluation of the Allometric Exponents in Prediction of Human Drug Clearance
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2014 Evaluation of the Allometric Exponents in Prediction of Human Drug Clearance Da Zhang Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Other Pharmacy and Pharmaceutical Sciences Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/3533 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. ©Da Zhang, 2014 All Rights Reserve EVALUATION OF THE ALLOMETRIC EXPONENTS IN PREDICTION OF HUMAN DRUG CLEARANCE A Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy at Virginia Commonwealth University By Da Zhang Master of Science, University of Arizona, 2004 Director: F. Douglas Boudinot, Ph.D. Professor, School of Pharmacy, VCU Virginia Commonwealth University Richmond, Virginia August, 2014 ACKNOWLEDGEMENTS First and foremost, I would like to sincerely thank my advisor, Dr. F. Douglas Boudinot, for giving me the opportunity to pursue graduate studies under his guidance and for his continuous professional support, guidance, encouragement and patience throughout my graduate program. He was always delighted in sharing his vast knowledge and kind warmth. I would also like to thank Dr. Ahmad for his scientific advice and support through the complet ion of my degree. He has been a kind and helpful mentor for me. I would like to acknowledge my graduate committee members, Drs. -

Predictive Value of Parkinsonian Primates in Pharmacological Studies, a Comparison Between the Macaque, Marmoset and Squirrel Monkey I
Downloaded from jpet.aspetjournals.org at ASPET Journals on October 1, 2021 NV PH JPET #247171 i HA,PH 1 Adjia Hamadjida, Philippe Huot Adjia Philippe Hamadjida, , JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. Published on March 9, 2018 as DOI: 10.1124/jpet.117.247171 JPET Fast Forward. -

(12) United States Patent (10) Patent N0.: US 7,964,607 B2 Verhoest Et A1
US007964607B2 (12) United States Patent (10) Patent N0.: US 7,964,607 B2 Verhoest et a1. (45) Date of Patent: Jun. 21, 2011 (54) PYRAZOLO[3,4-D]PYRIMIDINE FOREIGN PATENT DOCUMENTS COMPOUNDS EP 1460077 9/2004 WO 02085904 10/2002 (75) Inventors: Patrick Robert Verhoest, Old Lyme, CT WO 2004037176 5/2004 (US); Caroline ProulX-Lafrance, Ledyard, CT (US) OTHER PUBLICATIONS Wunder et a1, M01. PharmacoL, v01. 28, N0. 6, (2005), pp. 1776 (73) Assignee: P?zer Inc., New York, NY (U S) 1781. van der Staay et a1, Neuropharmacology, v01. 55 (2008), pp. 908 ( * ) Notice: Subject to any disclaimer, the term of this 918. patent is extended or adjusted under 35 USC 154(b) by 562 days. Primary Examiner * Susanna Moore (74) Attorney, Agent, or Firm * Jennifer A. Kispert; (21) Appl.No.: 12/118,062 Michael Herman (22) Filed: May 9, 2008 (57) ABSTRACT (65) Prior Publication Data The invention provides PDE9-inhibiting compounds of For US 2009/0030003 A1 Jan. 29, 2009 mula (I), Related US. Application Data (60) Provisional application No. 60/917,333, ?led on May 11, 2007. (51) Int. Cl. C07D 48 7/04 (2006.01) A61K 31/519 (2006.01) A61P 25/28 (2006.01) (52) US. Cl. ................................... .. 514/262.1; 544/262 (58) Field of Classi?cation Search ................ .. 544/262; 5 1 4/2 62 .1 See application ?le for complete search history. and pharmaceutically acceptable salts thereof, Wherein R, R1, (56) References Cited R2 and R3 are as de?ned herein. Pharmaceutical compositions containing the compounds of Formula I, and uses thereof in U.S. -

Cardiac Glycosides Provide Neuroprotection Against Ischemic Stroke: Discovery by a Brain Slice-Based Compound Screening Platform
Cardiac glycosides provide neuroprotection against ischemic stroke: Discovery by a brain slice-based compound screening platform James K. T. Wang*†, Stuart Portbury*‡, Mary Beth Thomas*§, Shawn Barney*, Daniel J. Ricca*, Dexter L. Morris*¶, David S. Warnerʈ, and Donald C. Lo*,**†† *Cogent Neuroscience, Inc., Durham, NC 27704; ʈMultidisciplinary Neuroprotection Laboratories and Department of Anesthesiology, Duke University Medical Center, Durham, NC 27710; and **Center for Drug Discovery and Department of Neurobiology, Duke University Medical Center, Durham, NC 27704 Edited by Charles F. Stevens, The Salk Institute for Biological Studies, La Jolla, CA, and approved May 17, 2006 (received for review February 3, 2006) We report here the results of a chemical genetic screen using small intrinsically problematic for a number of reasons, including inher- molecules with known pharmacologies coupled with a cortical ent limitations on therapeutic time window and clinically limiting brain slice-based model for ischemic stroke. We identified a small- side-effect profiles. Consequently, much attention has been focused molecule compound not previously appreciated to have neuropro- in recent years on using genomic, proteomic, and other systems tective action in ischemic stroke, the cardiac glycoside neriifolin, biology approaches in identifying new drug target candidates for and demonstrated that its properties in the brain slice assay stroke drug intervention (see review in ref. 5). included delayed therapeutic potential exceeding 6 h. Neriifolin is In this context we developed a tissue-based, high-content assay structurally related to the digitalis class of cardiac glycosides, and model for ischemic stroke based on biolistic transfection of visual ؉ ؉ its putative target is the Na ͞K -ATPase. -

United States Patent (10 ) Patent No.: US 10,660,887 B2 Javitt (45 ) Date of Patent: *May 26 , 2020
US010660887B2 United States Patent (10 ) Patent No.: US 10,660,887 B2 Javitt (45 ) Date of Patent : *May 26 , 2020 (54 ) COMPOSITION AND METHOD FOR (56 ) References Cited TREATMENT OF DEPRESSION AND PSYCHOSIS IN HUMANS U.S. PATENT DOCUMENTS 6,228,875 B1 5/2001 Tsai et al. ( 71 ) Applicant : Glytech , LLC , Ft. Lee , NJ (US ) 2004/0157926 A1 * 8/2004 Heresco - Levy A61K 31/198 514/561 (72 ) Inventor: Daniel C. Javitt , Ft. Lee , NJ (US ) 2005/0261340 Al 11/2005 Weiner 2006/0204486 Al 9/2006 Pyke et al . 2008/0194631 Al 8/2008 Trovero et al. ( 73 ) Assignee : GLYTECH , LLC , Ft. Lee , NJ (US ) 2008/0194698 A1 8/2008 Hermanussen et al . 2010/0069399 A1 * 3/2010 Gant CO7D 401/12 ( * ) Notice : Subject to any disclaimer, the term of this 514 / 253.07 patent is extended or adjusted under 35 2010/0216805 Al 8/2010 Barlow 2011/0207776 Al 8/2011 Buntinx U.S.C. 154 ( b ) by 95 days . 2011/0237602 A1 9/2011 Meltzer This patent is subject to a terminal dis 2011/0306586 Al 12/2011 Khan claimer . 2012/0041026 A1 2/2012 Waizumi ( 21 ) Appl. No.: 15 /650,912 FOREIGN PATENT DOCUMENTS CN 101090721 12/2007 ( 22 ) Filed : Jul. 16 , 17 KR 2007 0017136 2/2007 WO 2005/065308 7/2005 (65 ) Prior Publication Data WO 2005/079756 9/2005 WO 2011044089 4/2011 US 2017/0312275 A1 Nov. 2 , 2017 WO 2012/104852 8/2012 WO 2005/000216 9/2013 WO 2013138322 9/2013 Related U.S. Application Data (63 ) Continuation of application No. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -
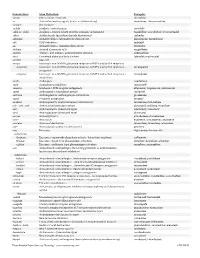
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

Supplemental Material Liu Page 1
Supplementary material Stroke Vasc Neurol Supplemental Material Liu Page 1 Supplemental Material Chinese Stroke Association Guidelines for Clinical Management of Cerebrovascular Disorders Executive Summary and 2019 Update of Clinical Management of Ischemic Cerebrovascular Diseases Liu L, et al. Stroke Vasc Neurol 2020; 5:e000378. doi: 10.1136/svn-2020-000378 Supplementary material Stroke Vasc Neurol Supplemental Material Liu Page 2 Section 1 Definitions associated with ischemic cerebrovascular diseases. The definitions of ischemic cerebrovascular disease are shown in Table 1. Section 2 Emergency assessment and diagnosis of ischemic stroke patients Please refer to Figure 1 for details of the management process for ischemic stroke patients in the acute phase. I. First assessment of emergency (I) Medical history collection Acute ischemic stroke (AIS) patients suffer from acute onset. It is very important to inquire about the time of occurrence of symptoms, onset characteristics and progress, including symptom persistence, fluctuations and relief. If the disease starts onset during sleep, the time when the patient's last normal performance should be recorded. These patients may also get sick shortly before waking up, and the onset time is uncertain, but clinically the final normal time should still be calculated as the last normal time [1-2]. Using MRI to screen wake-up stroke patients who are not suitable for thrombectomy, intravenous thrombolysis helps patients get good prognosis [3]. Before the occurrence of current symptoms, some patients will have similar TIA symptoms that can be relieved automatically. The time window for thrombolytic therapy is calculated based on the occurrence time of current symptoms. The inquiry about whether there are causes related to haemorrhagic stroke before the occurrence of symptoms, such as emotional excitement, intense exercise, sudden change of body position, excessive blood pressure reduction, as well as whether there is a history of Liu L, et al. -

Understanding History, and Not Repeating It. Neuroprotection For
Clinical Neurology and Neurosurgery 129 (2015) 1–9 Contents lists available at ScienceDirect Clinical Neurology and Neurosurgery jo urnal homepage: www.elsevier.com/locate/clineuro Review Understanding history, and not repeating it. Neuroprotection for acute ischemic stroke: From review to preview a a b b,c a,b,c,d,∗ Stephen Grupke , Jason Hall , Michael Dobbs , Gregory J. Bix , Justin F. Fraser a Department of Neurosurgery, University of Kentucky, Lexington, USA b Department of Neurology, University of Kentucky, Lexington, USA c Department of Anatomy and Neurobiology, University of Kentucky, Lexington, USA d Department of Radiology, University of Kentucky, Lexington, USA a r t i c l e i n f o a b s t r a c t Article history: Background: Neuroprotection for ischemic stroke is a growing field, built upon the elucidation of the Received 17 April 2014 biochemical pathways of ischemia first studied in the 1970s. Beginning in the early 1990s, means by Received in revised form 7 November 2014 which to pharmacologically intervene and counteract these pathways have been sought, though with Accepted 13 November 2014 little clinical success. Through a comprehensive review of translations from laboratory to clinic, we aim Available online 3 December 2014 to evaluate individual mechanisms of action, while highlighting potential barriers to success that will guide future research. Keywords: Methods: The MEDLINE database and The Internet Stroke Center clinical trials registry were queried Acute stroke Angiography for trials involving the use of neuroprotective agents in acute ischemic stroke in human subjects. For the purpose of the review, neuroprotective agents refer to medications used to preserve or protect the Brain ischemia Drug trials potentially ischemic tissue after an acute stroke, excluding treatments designed to re-establish perfusion.