FLT3 Signalling Pathways in Acute Myeloid Leukaemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human RET Kinase Protein (His Tag)
Human RET Kinase Protein (His Tag) Catalog Number: 11997-H08H1 General Information SDS-PAGE: Gene Name Synonym: CDHF12; CDHR16; HSCR1; MEN2A; MEN2B; MTC1; PTC; RET-ELE1; RET51 Protein Construction: A DNA sequence encoding the extracellular domain of human RET (P07949-1) (Met 1-Arg 635) was fused with a polyhistidine tag at the N- terminus. Source: Human Expression Host: HEK293 Cells QC Testing Purity: > 92 % as determined by SDS-PAGE Protein Description Endotoxin: RET proto-oncogene, also known as RET, is a cell-surface molecule that < 1.0 EU per μg of the protein as determined by the LAL method transduce signals for cell growth and differentiation. It contains 1 cadherin domain and 1 protein kinase domain. RET proto-oncogene belongs to the Stability: protein kinase superfamily, tyr protein kinase family. RET proto-oncogene is involved in numerous cellular mechanisms including cell proliferation, ℃ Samples are stable for up to twelve months from date of receipt at -70 neuronal navigation, cell migration, and cell differentiation upon binding with glial cell derived neurotrophic factor family ligands. It phosphorylates Leu 29 Predicted N terminal: PTK2/FAK1 and regulates both cell death/survival balance and positional Molecular Mass: information. RET is required for the molecular mechanisms orchestration during intestine organogenesis; involved in the development of enteric The recombinant human RET consists of 618 amino acids and has a nervous system and renal organogenesis during embryonic life; promotes calculated molecular mass of 69.1 kDa. The apparent molecular mass of the formation of Peyer's patch-like structures; modulates cell adhesion via the protein is approximately 110-120 kDa in SDS-PAGE under reducing its cleavage; involved in the development of the neural crest. -

Promoter Janus Kinase 3 Proximal Characterization and Analysis Of
The Journal of Immunology Characterization and Analysis of the Proximal Janus Kinase 3 Promoter1 Martin Aringer,2*† Sigrun R. Hofmann,2* David M. Frucht,* Min Chen,* Michael Centola,* Akio Morinobu,* Roberta Visconti,* Daniel L. Kastner,* Josef S. Smolen,† and John J. O’Shea3* Janus kinase 3 (Jak3) is a nonreceptor tyrosine kinase essential for signaling via cytokine receptors that comprise the common ␥-chain (␥c), i.e., the receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Jak3 is preferentially expressed in hemopoietic cells and is up-regulated upon cell differentiation and activation. Despite the importance of Jak3 in lymphoid development and immune function, the mechanisms that govern its expression have not been defined. To gain insight into this issue, we set out to characterize the Jak3 promoter. The 5-untranslated region of the Jak3 gene is interrupted by a 3515-bp intron. Upstream of this intron and the transcription initiation site, we identified an ϳ1-kb segment that exhibited lymphoid-specific promoter activity and was responsive to TCR signals. Truncation of this fragment revealed that core promoter activity resided in a 267-bp fragment that contains putative Sp-1, AP-1, Ets, Stat, and other binding sites. Mutation of the AP-1 sites significantly diminished, whereas mutation of the Ets sites abolished, the inducibility of the promoter construct. Chromatin immunoprecipitation assays showed that histone acetylation correlates with mRNA expression and that Ets-1/2 binds this region. Thus, transcription factors that bind these sites, especially Ets family members, are likely to be important regulators of Jak3 expression. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Calycosin Down-Regulates C-Met to Suppress Development of Glioblastomas
J Biosci (2019) 44:96 Ó Indian Academy of Sciences DOI: 10.1007/s12038-019-9904-4 (0123456789().,-volV)(0123456789().,-volV) Calycosin down-regulates c-Met to suppress development of glioblastomas , XIAOHU NIE ,YUE ZHOU* ,XIAOBING LI,JIE XU,XUYAN PAN,RUI YIN and BIN LU Department of Neurosurgery, Huzhou Central Hospital, Huzhou, Zhejiang, People’s Republic of China *Corresponding author (Email, [email protected]) These authors contributed equally to this work. MS received 15 October 2018; accepted 9 June 2019; published online 7 August 2019 The antitumor effect of calycosin has been widely studied, but the targets of calycosin against glioblastomas are still unclear. In this study we focused on revealing c-Met as a potential target of calycosin suppressing glioblastomas. In this study, suppressed-cell proliferation and cell invasion together with induced-cell apoptosis appeared in calycosin-treated U251 and U87 cells. Under treatment of calycosin, the mRNA expression levels of Dtk, c-Met, Lyn and PYK2 were observed in U87 cells. Meanwhile a western blot assay showed that c-Met together with matrix metalloproteinases-9 (MMP9) and phosphorylation of the serine/threonine kinase AKT (p-AKT) was significantly down-regulated by calycosin. Furthermore, overexpressed c-Met in U87 enhanced the expression level of MMP9 and p-AKT and also improved cell invasion. Additionally, the expression levels of c-Met, MMP9 and p-AKT were inhibited by calycosin in c-Met overex- pressed cells. However, an AKT inhibitor (LY294002) only effected on MMP9 and p-AKT, not on c-Met. These data collectively indicated that calycosin possibility targeting on c-Met and exert an anti-tumor role via MMP9 and AKT. -

JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: a Focus on the Present and an Outlook on the Future
biomolecules Review JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future 1, 2, , 3 1,4 Jacopo Angelini y , Rossella Talotta * y , Rossana Roncato , Giulia Fornasier , Giorgia Barbiero 1, Lisa Dal Cin 1, Serena Brancati 1 and Francesco Scaglione 5 1 Postgraduate School of Clinical Pharmacology and Toxicology, University of Milan, 20133 Milan, Italy; [email protected] (J.A.); [email protected] (G.F.); [email protected] (G.B.); [email protected] (L.D.C.); [email protected] (S.B.) 2 Department of Clinical and Experimental Medicine, Rheumatology Unit, AOU “Gaetano Martino”, University of Messina, 98100 Messina, Italy 3 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico di Aviano (CRO), Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Pordenone, 33081 Aviano, Italy; [email protected] 4 Pharmacy Unit, IRCCS-Burlo Garofolo di Trieste, 34137 Trieste, Italy 5 Head of Clinical Pharmacology and Toxicology Unit, Grande Ospedale Metropolitano Niguarda, Department of Oncology and Onco-Hematology, Director of Postgraduate School of Clinical Pharmacology and Toxicology, University of Milan, 20162 Milan, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-090-2111; Fax: +39-090-293-5162 Co-first authors. y Received: 16 May 2020; Accepted: 1 July 2020; Published: 5 July 2020 Abstract: Janus kinase inhibitors (JAKi) belong to a new class of oral targeted disease-modifying drugs which have recently revolutionized the therapeutic panorama of rheumatoid arthritis (RA) and other immune-mediated diseases, placing alongside or even replacing conventional and biological drugs. -

FLT3 Inhibitors in Acute Myeloid Leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2*
Wu et al. Journal of Hematology & Oncology (2018) 11:133 https://doi.org/10.1186/s13045-018-0675-4 REVIEW Open Access FLT3 inhibitors in acute myeloid leukemia Mei Wu1, Chuntuan Li2 and Xiongpeng Zhu2* Abstract FLT3 mutations are one of the most common findings in acute myeloid leukemia (AML). FLT3 inhibitors have been in active clinical development. Midostaurin as the first-in-class FLT3 inhibitor has been approved for treatment of patients with FLT3-mutated AML. In this review, we summarized the preclinical and clinical studies on new FLT3 inhibitors, including sorafenib, lestaurtinib, sunitinib, tandutinib, quizartinib, midostaurin, gilteritinib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG 925, TTT-3002, and FF-10101. New generation FLT3 inhibitors and combination therapies may overcome resistance to first-generation agents. Keywords: FMS-like tyrosine kinase 3 inhibitors, Acute myeloid leukemia, Midostaurin, FLT3 Introduction RAS, MEK, and PI3K/AKT pathways [10], and ultim- Acute myeloid leukemia (AML) remains a highly resist- ately causes suppression of apoptosis and differentiation ant disease to conventional chemotherapy, with a me- of leukemic cells, including dysregulation of leukemic dian survival of only 4 months for relapsed and/or cell proliferation [11]. refractory disease [1]. Molecular profiling by PCR and Multiple FLT3 inhibitors are in clinical trials for treat- next-generation sequencing has revealed a variety of re- ing patients with FLT3/ITD-mutated AML. In this re- current gene mutations [2–4]. New agents are rapidly view, we summarized the preclinical and clinical studies emerging as targeted therapy for high-risk AML [5, 6]. on new FLT3 inhibitors, including sorafenib, lestaurtinib, In 1996, FMS-like tyrosine kinase 3/internal tandem du- sunitinib, tandutinib, quizartinib, midostaurin, gilteriti- plication (FLT3/ITD) was first recognized as a frequently nib, crenolanib, cabozantinib, Sel24-B489, G-749, AMG mutated gene in AML [7]. -

Supplementary Materials
Supplementary Suppl. Figure 1: MAPK signalling pathway of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC-5. Suppl. Figure 2: Cell cycle pathway of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC- 5. Suppl. Figure 3: Cancer pathways of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC-5. Suppl. Figure 4: Phosphorylation level of A: ARAF, B: EPHA1, C: EPHA2, D: EPHA7 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 5: Phosphorylation Level of A: KIT, B: PTPN11, C: PIK3R1, D: PTPN6 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 6: Phosphorylation Level of A: KDR, B: EFS, C: AKT1, D: PTK2B/FAK2 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 7: Scoreplots and volcanoplots of PTK upstream kinase analysis: A: Scoreplot of PTK- Upstream kinase analysis for NCI-H2052 cells. B: Volcanoplot of PTK-Upstream kinase analysis for NCI-H2052 cells. C: Scoreplot of PTK-Upstream kinase analysis for NCI-H2452 cells. D: Volcanoplot of PTK-Upstream kinase analysis for NCI-H2452 cells. E: Scoreplot of PTK-Upstream kinase analysis for MSTO-211H cells. F: Volcanoplot of PTK-Upstream kinase analysis for MSTO- 211H cells. G: Scoreplot of PTK-Upstream kinase analysis for MRC-5cells. H: Volcanoplot of PTK- Upstream kinase analysis for MRC-5 cells. Suppl. Figure 8: Scoreplots and volcanoplots of STK upstream kinase analysis: A: Scoreplot of STK- Upstream kinase analysis for NCI-H2052 cells. -

Inhibition of DDR1-BCR Signalling by Nilotinib As a New Therapeutic
Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer Maya Jeitany, Cédric Leroy, Priscillia Tosti, Marie Lafitte, Jordy Le Guet, Valérie Simon, Debora Bonenfant, Bruno Robert, Fanny Grillet, Caroline Mollevi, et al. To cite this version: Maya Jeitany, Cédric Leroy, Priscillia Tosti, Marie Lafitte, Jordy Le Guet, et al.. Inhibition of DDR1- BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Molecular Medicine, Wiley Open Access, 2018, 10 (4), pp.e7918. 10.15252/emmm.201707918. hal- 01872978 HAL Id: hal-01872978 https://hal.archives-ouvertes.fr/hal-01872978 Submitted on 12 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Research Article Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer Maya Jeitany1,†, Cédric Leroy1,2,3,†, Priscillia Tosti1,†, Marie Lafitte1, Jordy Le Guet1, Valérie Simon1, Debora Bonenfant2, Bruno Robert4, Fanny Grillet5, Caroline Mollevi4, Safia El Messaoudi4, Amaëlle Otandault4, Lucile Canterel-Thouennon4, Muriel Busson4, Alain R Thierry4, Pierre Martineau4, Julie Pannequin5, Serge Roche1,*,† & Audrey Sirvent1,†,** Abstract The current clinical management involves surgical removal of the primary tumour, often associated with chemotherapy. -

Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling While Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells
Published OnlineFirst February 14, 2013; DOI: 10.1158/1535-7163.MCT-12-0305 Molecular Cancer Cancer Therapeutics Insights Therapeutics Transient Exposure to Quizartinib Mediates Sustained Inhibition of FLT3 Signaling while Specifically Inducing Apoptosis in FLT3-Activated Leukemia Cells Ruwanthi N. Gunawardane, Ronald R. Nepomuceno, Allison M. Rooks, Jeremy P. Hunt, Jill M. Ricono, Barbara Belli, and Robert C. Armstrong Abstract Fms-like tyrosine kinase 3 (FLT3) is implicated in the pathogenesis of acute myeloid leukemia (AML). FLT3-activating internal tandem duplication (ITD) mutations are found in approximately 30% of patients with AML and are associated with poor outcome in this patient population. Quizartinib (AC220) has previously been shown to be a potent and selective FLT3 inhibitor. In the current study, we expand on previous observations by showing that quizartinib potently inhibits the phosphorylation of FLT3 and downstream signaling molecules independent of FLT3 genotype, yet induces loss of viability only in cells expressing constitutively activated FLT3. We further show that transient exposure to quizartinib, whether in vitro or in vivo, leads to prolonged inhibition of FLT3 signaling, induction of apoptosis, and drastic reductions in tumor volume and pharmacodynamic endpoints. In vitro experiments suggest that these prolonged effects are mediated by slow binding kinetics that provide for durable inhibition of the kinase following drug removal/clearance. Together these data suggest quizartinib, with its unique combination of selectivity and potent/sustained inhibition of FLT3, may provide a safe and effective treatment against FLT3-driven leukemia. Mol Cancer Ther; 12(4); 1–10. Ó2013 AACR. Introduction downstream signaling cascades including the Ras/ Fms-like tyrosine kinase 3 (FLT3) is a member of the MAPK, PI3K/Akt, and STAT5 pathways (1, 9–13). -

Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia
Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia Title Page by Ravi K. Patel Bachelor of Science, University of Minnesota, 2013 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2019 Commi ttee Membership Pa UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE Commi ttee Membership Page This dissertation was presented by Ravi K. Patel It was defended on May 31, 2019 and approved by Qiming (Jane) Wang, Associate Professor Pharmacology and Chemical Biology Vaughn S. Cooper, Professor of Microbiology and Molecular Genetics Adrian Lee, Professor of Pharmacology and Chemical Biology Laura Stabile, Research Associate Professor of Pharmacology and Chemical Biology Thomas E. Smithgall, Dissertation Director, Professor and Chair of Microbiology and Molecular Genetics ii Copyright © by Ravi K. Patel 2019 iii Abstract Src-Family Kinases Play an Important Role in Flt3-ITD Acute Myeloid Leukemia Prognosis and Drug Efficacy Ravi K. Patel, PhD University of Pittsburgh, 2019 Abstract Acute myelogenous leukemia (AML) is a disease characterized by undifferentiated bone-marrow progenitor cells dominating the bone marrow. Currently the five-year survival rate for AML patients is 27.4 percent. Meanwhile the standard of care for most AML patients has not changed for nearly 50 years. We now know that AML is a genetically heterogeneous disease and therefore it is unlikely that all AML patients will respond to therapy the same way. Upregulation of protein-tyrosine kinase signaling pathways is one common feature of some AML tumors, offering opportunities for targeted therapy. -
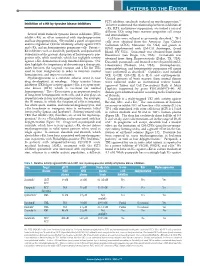
Letters to the Editor
LETTERS TO THE EDITOR FLT3 inhibitor, sorafenib, induced no myelosuppression.5,6 Inhibition of c-Kit by tyrosine kinase inhibitors To better understand the relationship between inhibition of c-Kit, FLT3, and marrow suppression, we studied a series of different TKIs using bone marrow progenitor cell assays Several small molecule tyrosine kinase inhibitors (TKIs) and immunoblots. inhibit c-Kit, an effect associated with myelosuppression Cell lines were cultured as previously described.2 TF-1 and hair depigmentation. We studied a panel of approved cells were obtained from the American Type Culture and investigational TKIs for inhibitory activity against FLT3 Collection (ATCC; Manassas, VA, USA) and grown in and c-Kit, and on hematopoietic progenitor cells. Potent c- RPMI supplemented with GM-CSF (Invitrogen, Grand Kit inhibitors such as dasatinib, pazopanib, and quizartinib Island, NY, USA). Quizartinib was obtained from Ambit demonstrated the greatest disruption of hematopoietic pro- Biosciences (San Diego, CA, USA). Crenolanib was genitor cells, while sorafenib, which has negligible activity obtained from Arog Pharmaceuticals (Dallas, TX, USA). against c-Kit, demonstrated only minimal disruption. Our Dasatinib, pazopanib, and imatinib were obtained from LC data highlight the importance of determining a therapeutic Laboratories (Woburn, MA, USA). Electrophoresis, index between the targeted receptor and c-Kit for TKIs immunoblotting, and hematopoietic progenitor cell assays used to treat malignancies in order to maintain normal were performed as described.2 Cytokines used included hematopoiesis and improve outcomes. SCF, G-CSF, GM-CSF, IL-3, IL-6, and erythropoietin. Myelosuppression is a common adverse event in new Unused portions of bone marrow from normal donors drug development in oncology. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal