Neoreviews-20.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of Arylsulfatase E Mutations, Functional Analysis of Novel Missense Alleles, and Determination of Potential Phenocopies
ORIGINAL RESEARCH ARTICLE © American College of Medical Genetics and Genomics A prospective study of brachytelephalangic chondrodysplasia punctata: identification of arylsulfatase E mutations, functional analysis of novel missense alleles, and determination of potential phenocopies Claudia Matos-Miranda, MSc, MD1, Graeme Nimmo, MSc1, Bradley Williams, MGC, CGC2, Carolyn Tysoe, PhD3, Martina Owens, BS3, Sherri Bale, PhD2 and Nancy Braverman, MS, MD1,4 Purpose: The only known genetic cause of brachytelephalangic Results: In this study, 58% of males had ARSE mutations. All mutant chondrodysplasia punctata is X-linked chondrodysplasia punctata 1 alleles had negligible arylsulfatase E activity. There were no obvi- (CDPX1), which results from a deficiency of arylsulfatase E (ARSE). ous genotype–phenotype correlations. Maternal etiologies were not Historically, ARSE mutations have been identified in only 50% of reported in most patients. male patients, and it was proposed that the remainder might rep- resent phenocopies due to maternal–fetal vitamin K deficiency and Conclusion: CDPX1 is caused by loss of arylsulfatase E activ- maternal autoimmune diseases. ity. Around 40% of male patients with brachytelephalangic chon- drodysplasia punctata do not have detectable ARSE mutations or Methods: To further evaluate causes of brachytelephalangic chon- known maternal etiological factors. Improved understanding of drodysplasia punctata, we established a Collaboration Education and arylsulfatase E function is predicted to illuminate other etiologies for Test Translation program for CDPX1 from 2008 to 2010. Of the 29 brachytelephalangic chondrodysplasia punctata. male probands identified, 17 had ARSE mutations that included 10 novel missense alleles and one single-codon deletion. To determine Genet Med 2013:15(8):650–657 pathogenicity of these and additional missense alleles, we transiently expressed them in COS cells and measured arylsulfatase E activity Key Words: arylsulfatase E; brachytelephalangic chondrodysplasia using the artificial substrate, 4-methylumbelliferyl sulfate. -

Esidencyofficial Publication of the Residents/Fellows Committee, American Academy of Dermatology Keeping Financially Afloat During Residency by Dean Monti
irinections D Fall 2016 ResidencyOfficial Publication of the Residents/Fellows Committee, American Academy of Dermatology Keeping financially afloat during residency By Dean Monti Resident lives are saturated with books, charts, and a considerable amount of study- ing. With boards and the rest of their careers looming ahead, there is obviously a lot of associated stress. One area of education that’s generally not covered in residency pro- grams, however, is debt. Directions recently reached out to residents about this topic, and received plenty of feedback regarding their concerns, fears, advice, and more. One resident we talked to, Jeffrey Kushner, DO — a PGY-3 at Saint Joseph Mercy Health System in Ann Arbor, Michigan — has been augmenting his studies with a strong inter- est in investment and personal finance. He has researched and read extensively on a variety of financial topics that dermatology residents face early in their careers, and has discovered some universal principals that all don’t realize that they’re lacking the skills 2016 discussed the staggering additional dermatology residents should be aware of. or knowledge to handle their debt until it expense of simply applying to dermatology He has subsequently lectured and given pre- becomes a reality. residency! sentations to his fellow residents on finance For most of us, having a growing moun- Fortunately, not everything is doom and and investment topics. He has no conflict of tain of debt is an anxiety-provoking issue, gloom. We as dermatologists are still well- interest — which is often a concern for those but it can lead some to take an “out of sight, compensated compared to our colleagues seeking advice. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Blueprint Genetics Comprehensive Growth Disorders / Skeletal
Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders Panel Test code: MA4301 Is a 374 gene panel that includes assessment of non-coding variants. This panel covers the majority of the genes listed in the Nosology 2015 (PMID: 26394607) and all genes in our Malformation category that cause growth retardation, short stature or skeletal dysplasia and is therefore a powerful diagnostic tool. It is ideal for patients suspected to have a syndromic or an isolated growth disorder or a skeletal dysplasia. About Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders This panel covers a broad spectrum of diseases associated with growth retardation, short stature or skeletal dysplasia. Many of these conditions have overlapping features which can make clinical diagnosis a challenge. Genetic diagnostics is therefore the most efficient way to subtype the diseases and enable individualized treatment and management decisions. Moreover, detection of causative mutations establishes the mode of inheritance in the family which is essential for informed genetic counseling. For additional information regarding the conditions tested on this panel, please refer to the National Organization for Rare Disorders and / or GeneReviews. Availability 4 weeks Gene Set Description Genes in the Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACAN# Spondyloepimetaphyseal dysplasia, aggrecan type, AD/AR 20 56 Spondyloepiphyseal dysplasia, Kimberley -

Blueprint Genetics Comprehensive Skeletal Dysplasias and Disorders
Comprehensive Skeletal Dysplasias and Disorders Panel Test code: MA3301 Is a 251 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of disorders involving the skeletal system. About Comprehensive Skeletal Dysplasias and Disorders This panel covers a broad spectrum of skeletal disorders including common and rare skeletal dysplasias (eg. achondroplasia, COL2A1 related dysplasias, diastrophic dysplasia, various types of spondylo-metaphyseal dysplasias), various ciliopathies with skeletal involvement (eg. short rib-polydactylies, asphyxiating thoracic dysplasia dysplasias and Ellis-van Creveld syndrome), various subtypes of osteogenesis imperfecta, campomelic dysplasia, slender bone dysplasias, dysplasias with multiple joint dislocations, chondrodysplasia punctata group of disorders, neonatal osteosclerotic dysplasias, osteopetrosis and related disorders, abnormal mineralization group of disorders (eg hypopohosphatasia), osteolysis group of disorders, disorders with disorganized development of skeletal components, overgrowth syndromes with skeletal involvement, craniosynostosis syndromes, dysostoses with predominant craniofacial involvement, dysostoses with predominant vertebral involvement, patellar dysostoses, brachydactylies, some disorders with limb hypoplasia-reduction defects, ectrodactyly with and without other manifestations, polydactyly-syndactyly-triphalangism group of disorders, and disorders with defects in joint formation and synostoses. Availability 4 weeks Gene Set Description -

Syndrome of the Month J Med Genet: First Published As 10.1136/Jmg.33.11.957 on 1 November 1996
J Med Genet 1996;33:957-961 957 Syndrome of the month J Med Genet: first published as 10.1136/jmg.33.11.957 on 1 November 1996. Downloaded from Achondrogenesis type 1B Andrea Superti-Furga Historical notes which tried to provide a quantitative basis (the In 1952, the name achondrogenesis (Greek for "femoral cylinder index") for qualitative "not producing cartilage") was given by Marco changes, did not prove helpful and was later Fraccaro, a young Italian pathologist (later to abandoned. In the late 1980s, it was shown become a well known cytogeneticist), to the that achondrogenesis type II was caused by condition he observed in a stillborn female structural mutations in collagen II and thus with severe micromelia and marked histological constituted the severe end of the spectrum of changes of cartilage.' Fraccaro noted a similar the collagen II chondrodysplasias.l'" case published by Parenti in 1936.2 Fraccaro's Borochowitz et al'4 provided convincing report (written in Italian) came to the know- histological criteria for the further subdivision ledge ofHans Grebe, who in 1939 had observed of achondrogenesis type I into IA (with ap- sisters (aged 7 and 11 years, born to con- parently normal cartilage matrix but inclusions sanguineous parents from the Black Forest re- in chondrocytes) and IB (with abnormal car- gion of Germany) with markedly short limbs tilage matrix; see below). These findings were and digits but normal trunk. Grebe became confirmed by another group shortly there- convinced that his patients were affected by after.'5 Using -
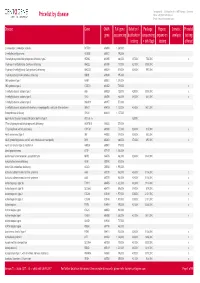
Pricelist by Disease Phone: +49 (0)381 203 652 222 E-Mail: [email protected]
Centogene AG :: Schillingallee 68 :: 18057 Rostock :: Germany Pricelist by disease Phone: +49 (0)381 203 652 222 E-mail: [email protected] Disease Gene OMIM Full gene Deletion/ Package Repeat Somatic Prenatal gene sequencing duplication (sequencing expansion analysis testing testing + del/dup) testing offered 2-aminoadipic 2-oxoadipic aciduria DHTKD1 614984 1.260,00 € 2-methylbutyrylglycinuria ACADSB 600301 796,00 € 3-beta-hydroxysteroid dehydrogenase deficiency type 2 HSD3B2 613890 468,00 € 437,00 € 755,00 € x 3-hydroxy-3-methylglutaryl-CoA lyase deficiency HMGCL 613898 737,00 € 421,00 € 1.008,00 € 3-hydroxy-3-methylglutaryl-CoA synthase 2 deficiency HMGCS2 600234 819,00 € 328,00 € 997,00 € 3-hydroxyisobutryl-CoA hydrolase deficiency HIBCH 610690 995,00 € 3MC syndrome type 1 MASP1 600521 1.278,00 € 3MC syndrome type 2 COLEC11 612502 729,00 € x 3-methylglutaconic aciduria type 1 AUH 600529 729,00 € 429,00 € 1.008,00 € x 3-methylglutaconic aciduria type 3 OPA3 606580 468,00 € 343,00 € 661,00 € x 3-methylglutaconic aciduria type 5 DNAJC19 608977 573,00 € 3-methylglutaconic aciduria with deafness, encephalopathy, and Leigh-like syndrome SERAC1 614725 1.127,00 € 424,00 € 1.401,00 € 5-oxoprolinase deficiency OPLAH 614243 1.127,00 € 6q24-related transient neonatal diabetes mellitus type 1 UPD chr. 6 328,00 € 17-beta hydroxysteroid dehydrogenase X deficiency HSD17B10 300256 573,00 € 17-hydroxylation activity deficiency CYP17A1 609300 737,00 € 328,00 € 915,00 € x 46,XX sex reversal type 1 SRY 480000 374,00 € 328,00 € 552,00 € 46,XY gonadal -

Skeletal Dysplasias Precision Panel Overview Indications
Skeletal Dysplasias Precision Panel Overview Skeletal Dysplasias, also known as osteochondrodysplasias, are a clinically and phenotypically heterogeneous group of more than 450 inherited disorders characterized by abnormalities mainly of cartilage and bone growth although they can also affect muscle, tendons and ligaments, resulting in abnormal shape and size of the skeleton and disproportion of long bones, spine and head. They differ in natural histories, prognoses, inheritance patterns and physiopathologic mechanisms. They range in severity from those that are embryonically lethal to those with minimum morbidity. Approximately 5% of children with congenital birth defects have skeletal dysplasias. Until recently, the diagnosis of skeletal dysplasia relied almost exclusively on careful phenotyping, however, the advent of genomic tests has the potential to make a more accurate and definite diagnosis based on the suspected clinical diagnosis. The 4 most common skeletal dysplasias are thanatophoric dysplasia, achondroplasia, osteogenesis imperfecta and achondrogenesis. The inheritance pattern of skeletal dysplasias is variable and includes autosomal dominant, recessive and X-linked. The Igenomix Skeletal Dysplasias Precision Panel can be used to make a directed and accurate differential diagnosis of skeletal abnormalities ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Mouse Models of Human Disease. Part II: Recent Progress and Future Directions
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Mouse models of human disease. Part II: Recent progress and future directions Mary A. Bedell, 1 David A. Largaespada, 2 Nancy A. Jenkins, and Neal G. Copeland 3 Mammalian Genetics Laboratory, ABL-Basic Research Program, NCI-Frederick Cancer Research and Development Center, Frederick, Maryland 21702-1201 USA The development of new methods for manipulating the the recent progress in this area. Throughout the text and mouse genome, including transgenic and embryonic tables, we have cited only the most recent papers and stem (ES) cell knockout technology, combined with refer to reviews whenever possible. Interested readers are greatly improved genetic and physical maps for mouse encouraged to read the primary papers on each model has revolutionized our ability to generate new mouse and associated disease. models of human disease. In Part I of this review (Bedell et al., this issue), we described in detail the various tech- Disorders of neural crest derivatives niques and genetic resources that have facilitated mouse model development. In Part II of this review we highlight Cells from the neural crest differentiate into many dif- some of the recent progress that has been made in mouse ferent cell types including melanocytes of the skin and model development and discuss areas where these inner ear, neuronal and glial components of the periph- mouse models are likely to contribute in the future. We eral nervous system, neuroendocrine cells of the adrenal have focused in part II only on those models where the medulla and thyroid, and cartilaginous and membranous homologous gene is mutated in both the human and bones of the skull. -

A Case of Achondrogenesis Type 2/ Hypochondrogenesis Due to Ade
J Clin Res Pediatr Endocrinol 2015;7(Suppl 2):77-92 A Case of Achondrogenesis Type 2/ Seven Cases of Williams-Beuren Syndrome: Hypochondrogenesis Due to a De Novo Mutation Endocrine Evaluation and Long-Term Follow-Up in the COL2A1 Gene Ayla Güven Gönül Çatlı1, Ayhan Abacı1, Ahmet Anık1, Ranad Shaneen2, Hale Ünver Tuhan1, Medeniyet University, Faculty of Medicine Göztepe Education Derya Erçal3, Ece Böber1, Niema A. Ibrahim2, and Research Hospital, Clinic of Pediatric Endocrinology, İstanbul, Turkey Mais O. Hashem2, Fowzan Sami Alkuraya2 Objectives: Hypercalcemia, hypothyroidism, and early 1 Dokuz Eylül University Faculty of Medicine, Department of puberty are the most common endocrine disorders defined Pediatric Endocrinology, İzmir, Turkey in Williams-Beuren syndrome (WBS). Here, the endocrine 2King Faisal Specialist Hospital and Research Center, Clinic of Genetics, Riyadh, Saudi Arabia evaluation and long-term follow-up of seven patients with 3Dokuz Eylül University Faculty of Medicine, Department of WBS are given. Pediatric Genetics, İzmir, Turkey Methods: Data were obtained from patients’ medical records. WBS was diagnosed by demonstration of the Achondrogenesis type 2/hypochondrogenesis is a rare deletion on chromosome 7 by using FISH method (7q11.23). cause of micromelic dwarfism in the neonatal period. Anthropometric measurements and growth velocities Severe short stature, narrow chest, increased lordosis, (GV) of patients during L-thyroxine (L-T4) treatment were protuberant abdomen, and characteristic facies are the evaluated. Thyroid ultrasonography was performed and typical clinical findings. A 13-month-old boy was referred to thyroid volume was calculated. L-T4 was started in patients our clinic for severe short stature and dysmorphic features. -

Risk Factors for and Clinical Management of Venous Thromboembolism During Pregnancy
Risk Factors for and Clinical Management of Venous Thromboembolism During Pregnancy Marissa D. Rybstein, MD, and Maria T. DeSancho, MD, MSc Dr Rybstein is a hematology-oncology Abstract: Venous thromboembolism (VTE), which comprises deep fellow and Dr DeSancho is an associ- vein thrombosis and pulmonary embolism, is one of the leading ate professor of clinical medicine in causes of non-obstetric maternal death in the United States. Physi- the Division of Hematology-Oncology ologic and anatomic changes associated with pregnancy set the of the Department of Medicine at New York-Presbyterian/Weill Cornell stage for a hypercoagulable state. In addition, other risk factors— Medical Center in New York, New including those associated with certain fetal characteristics such York. as low birth weight or stillbirth—have been correlated with an increased risk for VTE. Women with a personal or strong family history of VTE, as well as documented thrombophilia, represent Corresponding author: a unique group in whom antepartum and/or postpartum prophy- Maria T. DeSancho, MD, MSc Division of Hematology-Oncology laxis can be considered. The choice of anticoagulant therapy for Department of Medicine either treatment or prophylaxis in most cases is heparin, most New York-Presbyterian/Weill Cornell commonly low-molecular-weight heparin. This is owing to the fact Medical Center that vitamin K antagonists and the direct oral anticoagulants are 1305 York Avenue, 7th Floor, contraindicated in pregnancy because of potential teratogenicity. Room 51 With careful management and vigilant monitoring, appropriate New York, NY 10021 Tel: (646) 962-2065 anticoagulation can be used safely and effectively to improve Fax: (646) 962-1604 patient outcomes.