University of Birmingham a New Dawn for Androgens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

The Adrenal Androgen Precursors DHEA and Androstenedione (A4)
PERIPHERAL METABOLISM OF THE ADRENAL STEROID 11Β- HYDROXYANDROSTENEDIONE YIELDS THE POTENT ANDROGENS 11KETO- TESTOSTERONE AND 11KETO-DIHYDROTESTOSTERONE Elzette Pretorius1, Donita J Africander1, Maré Vlok2, Meghan S Perkins1, Jonathan Quanson1 and Karl-Heinz Storbeck1 1University of Stellenbosch, Department of Biochemistry, Stellenbosch, South Africa 2University of Stellenbosch, Mass Spectrometry Unit, Stellenbosch, South Africa The adrenal androgen precursors DHEA and androstenedione (A4) play an important role in the development and progression of castration resistant prostate cancer (CRPC) as they are converted to dihydrotestosterone (DHT) by steroidogenic enzymes expressed in CRPC tissue. We have recently shown that the adrenal C19 steroid 11β-hydroxyandrostenedione (11OHA4) serves as a precursor to the androgens, 11-ketotestosterone (11KT) and 11-keto-5α-dihydrotestosterone (11KDHT), and that the latter two steroids could play a role in CRPC. The aim of this study was therefore to characterise 11KT and 11KDHT in terms of their androgenic activity. Competitive whole cell binding assays revealed that 11KT and 11KDHT bind to the human androgen receptor (AR) with affinities similar to that of testosterone (T) and DHT. Transactivation assays on a synthetic androgen response element (ARE) demonstrated that the potencies and efficacies of 11KT and 11KDHT are comparable to that of T and DHT, respectively. Moreover, we show that both 11KT and 11KDHT induce the expression of AR-regulated genes (KLK3, TMPRSS2 and FKBP5) and cellular proliferation in the androgen dependent prostate cancer cell lines, LNCaP and VCaP. In most cases, 11KT and 11KDHT upregulated AR-regulated gene expression and increased LNCaP cell growth to a significantly higher extent than T and DHT. Mass spectrometry-based proteomics revealed that 11KT and 11KDHT, like T and DHT, results in the upregulation of multiple AR-regulated proteins in VCaP cells, with 11KDHT regulating more AR-regulated proteins than DHT. -

Effects on Steroid 5-Alpha Reductase Gene Expression of Thai Rice Bran Extracts and Molecular Dynamics Study on SRD5A2
biology Article Effects on Steroid 5-Alpha Reductase Gene Expression of Thai Rice Bran Extracts and Molecular Dynamics Study on SRD5A2 Chiranan Khantham 1 , Wipawadee Yooin 1,2 , Korawan Sringarm 2,3 , Sarana Rose Sommano 2,4 , Supat Jiranusornkul 1, Francisco David Carmona 5,6 , Wutigri Nimlamool 7 , Pensak Jantrawut 1,2, Pornchai Rachtanapun 8,9 and Warintorn Ruksiriwanich 1,2,8,* 1 Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand; [email protected] (C.K.); [email protected] (W.Y.); [email protected] (S.J.); [email protected] (P.J.) 2 Cluster of Research and Development of Pharmaceutical and Natural Products Innovation for Human or Animal, Chiang Mai University, Chiang Mai 50200, Thailand; [email protected] (K.S.); [email protected] (S.R.S.) 3 Department of Animal and Aquatic Sciences, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 4 Plant Bioactive Compound Laboratory (BAC), Department of Plant and Soil Sciences, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 5 Departamento de Genética e Instituto de Biotecnología, Universidad de Granada, 18071 Granada, Spain; [email protected] 6 Instituto de Investigación Biosanitaria ibs.GRANADA, 18014 Granada, Spain 7 Citation: Khantham, C.; Yooin, W.; Department of Pharmacology, Faculty of Medicine, Chiang Mai University, Chiang Mai 50200, Thailand; Sringarm, K.; Sommano, S.R.; [email protected] 8 Cluster of Agro Bio-Circular-Green Industry, Faculty of Agro-Industry, Chiang Mai University, Jiranusornkul, S.; Carmona, F.D.; Chiang Mai 50100, Thailand; [email protected] Nimlamool, W.; Jantrawut, P.; 9 School of Agro-Industry, Faculty of Agro-Industry, Chiang Mai University, Chiang Mai 50100, Thailand Rachtanapun, P.; Ruksiriwanich, W. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Pharmacology/Therapeutics II Block III Lectures 2013-14
Pharmacology/Therapeutics II Block III Lectures 2013‐14 66. Hypothalamic/pituitary Hormones ‐ Rana 67. Estrogens and Progesterone I ‐ Rana 68. Estrogens and Progesterone II ‐ Rana 69. Androgens ‐ Rana 70. Thyroid/Anti‐Thyroid Drugs – Patel 71. Calcium Metabolism – Patel 72. Adrenocorticosterioids and Antagonists – Clipstone 73. Diabetes Drugs I – Clipstone 74. Diabetes Drugs II ‐ Clipstone Pharmacology & Therapeutics Neuroendocrine Pharmacology: Hypothalamic and Pituitary Hormones, March 20, 2014 Lecture Ajay Rana, Ph.D. Neuroendocrine Pharmacology: Hypothalamic and Pituitary Hormones Date: Thursday, March 20, 2014-8:30 AM Reading Assignment: Katzung, Chapter 37 Key Concepts and Learning Objectives To review the physiology of neuroendocrine regulation To discuss the use neuroendocrine agents for the treatment of representative neuroendocrine disorders: growth hormone deficiency/excess, infertility, hyperprolactinemia Drugs discussed Growth Hormone Deficiency: . Recombinant hGH . Synthetic GHRH, Recombinant IGF-1 Growth Hormone Excess: . Somatostatin analogue . GH receptor antagonist . Dopamine receptor agonist Infertility and other endocrine related disorders: . Human menopausal and recombinant gonadotropins . GnRH agonists as activators . GnRH agonists as inhibitors . GnRH receptor antagonists Hyperprolactinemia: . Dopamine receptor agonists 1 Pharmacology & Therapeutics Neuroendocrine Pharmacology: Hypothalamic and Pituitary Hormones, March 20, 2014 Lecture Ajay Rana, Ph.D. 1. Overview of Neuroendocrine Systems The neuroendocrine -

DHEA Sulfate, and Aging: Contribution of the Dheage Study to a Sociobiomedical Issue
Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue Etienne-Emile Baulieua,b, Guy Thomasc, Sylvie Legraind, Najiba Lahloue, Marc Rogere, Brigitte Debuiref, Veronique Faucounaug, Laurence Girardh, Marie-Pierre Hervyi, Florence Latourj, Marie-Ce´ line Leaudk, Amina Mokranel, He´ le` ne Pitti-Ferrandim, Christophe Trivallef, Olivier de Lacharrie` ren, Stephanie Nouveaun, Brigitte Rakoto-Arisono, Jean-Claude Souberbiellep, Jocelyne Raisonq, Yves Le Boucr, Agathe Raynaudr, Xavier Girerdq, and Franc¸oise Foretteg,j aInstitut National de la Sante´et de la Recherche Me´dicale Unit 488 and Colle`ge de France, 94276 Le Kremlin-Biceˆtre, France; cInstitut National de la Sante´et de la Recherche Me´dicale Unit 444, Hoˆpital Saint-Antoine, 75012 Paris, France; dHoˆpital Bichat, 75877 Paris, France; eHoˆpital Saint-Vincent de Paul, 75014 Paris, France; fHoˆpital Paul Brousse, 94804 Villejuif, France; gFondation Nationale de Ge´rontologie, 75016 Paris, France; hHoˆpital Charles Foix, 94205 Ivry, France; iHoˆpital de Biceˆtre, 94275 Biceˆtre, France; jHoˆpital Broca, 75013 Paris, France; kCentre Jack-Senet, 75015 Paris, France; lHoˆpital Sainte-Perine, 75016 Paris, France; mObservatoire de l’Age, 75017 Paris, France; nL’Ore´al, 92583 Clichy, France; oInstitut de Sexologie, 75116 Paris, France; pHoˆpital Necker, 75015 Paris, France; qHoˆpital Broussais, 75014 Paris, France; and rHoˆpital Trousseau, 75012 Paris, France Contributed by Etienne-Emile Baulieu, December 23, 1999 The secretion and the blood levels of the adrenal steroid dehydro- number of consumers. Extravagant publicity based on fantasy epiandrosterone (DHEA) and its sulfate ester (DHEAS) decrease pro- (‘‘fountain of youth,’’ ‘‘miracle pill’’) or pseudoscientific asser- foundly with age, and the question is posed whether administration tion (‘‘mother hormone,’’ ‘‘antidote for aging’’) has led to of the steroid to compensate for the decline counteracts defects unfounded radical assertions, from superactivity (‘‘keep young,’’ associated with aging. -

Characterization of Precursor-Dependent Steroidogenesis in Human Prostate Cancer Models
cancers Article Characterization of Precursor-Dependent Steroidogenesis in Human Prostate Cancer Models Subrata Deb 1 , Steven Pham 2, Dong-Sheng Ming 2, Mei Yieng Chin 2, Hans Adomat 2, Antonio Hurtado-Coll 2, Martin E. Gleave 2,3 and Emma S. Tomlinson Guns 2,3,* 1 Department of Pharmaceutical Sciences, College of Pharmacy, Larkin University, Miami, FL 33169, USA; [email protected] 2 The Vancouver Prostate Centre at Vancouver General Hospital, 2660 Oak Street, Vancouver, BC V6H 3Z6, Canada; [email protected] (S.P.); [email protected] (D.-S.M.); [email protected] (M.Y.C.); [email protected] (H.A.); [email protected] (A.H.-C.); [email protected] (M.E.G.) 3 Department of Urologic Sciences, Faculty of Medicine, University of British Columbia, Vancouver, BC V5Z 1M9, Canada * Correspondence: [email protected]; Tel.: +1-604-875-4818 Received: 14 August 2018; Accepted: 17 September 2018; Published: 20 September 2018 Abstract: Castration-resistant prostate tumors acquire the independent capacity to generate androgens by upregulating steroidogenic enzymes or using steroid precursors produced by the adrenal glands for continued growth and sustainability. The formation of steroids was measured by liquid chromatography-mass spectrometry in LNCaP and 22Rv1 prostate cancer cells, and in human prostate tissues, following incubation with steroid precursors (22-OH-cholesterol, pregnenolone, 17-OH-pregnenolone, progesterone, 17-OH-progesterone). Pregnenolone, progesterone, 17-OH-pregnenolone, and 17-OH-progesterone increased C21 steroid (5-pregnan-3,20-dione, 5-pregnan-3,17-diol-20-one, 5-pregnan-3-ol-20-one) formation in the backdoor pathway, and demonstrated a trend of stimulating dihydroepiandrosterone or its precursors in the backdoor pathway in LNCaP and 22Rv1 cells. -

Role of Transport Systems in Cortisol Release from Human Adrenal Cells
Role of transport systems in cortisol release from human adrenal cells Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultäten der Georg-August-Universität zu Göttingen vorgelegt von Abdul Rahman Asif aus Gujrat, Pakistan Göttingen 2004 D7 Referent: Prof. Dr. R. Hardeland Korreferent: Prof. Dr. D. Gradmann Tag der mündlichen Prüfung: To My parents & in loving memory of my grandma! She could not wait! CONTENTS I ABSTRACT ................................................................................................... IV LIST OF ABBREVIATIONS .......................................................................... VI 1 INTRODUCTION........................................................................................1 1.1 THE ADRENAL GLAND ANATOMY ...............................................................................................1 1.2 ADRENAL GLAND HORMONES....................................................................................................2 1.2.1 Biosynthesis of the steroid hormones...........................................................................................2 1.2.2 Regulation of adrenal glands........................................................................................................6 1.2.3 Actions of adrenal steroids ...........................................................................................................7 1.3 HUMAN ADRENOCORTICAL CELLS ............................................................................................8 -

Steroidogenic Pathway Poster-COMPLEX-24X36
Cholesterol The Steroidogenic Pathways Congenital adrenal hyperplasia (CAH), Hyperglycemia, azole antifungals, aging, smoking, E2, progesterone, azole smoking, dioxin toxicity, HIV, licorice, Phytoestrogens, flaxseed, licorice, Alcohol, IL-6, abdominal obesity, DHEA, antifungals, spironolactone etomidate, liarozole progestins, tamoxifen forskolin (-) (-) (-) (+) Pregnenolone CYPc17 17-OH-Pregnenolone 17,20 Lyase DHEAA17βHSD ndrostenediol (+) Hyperglycemia, hyperinsulinemia, PCOS (ovary), stress, alcohol, antiepileptics (+) CAH, chronic ETOH ingestion, PCB toxicity, progestins, D D D (-) (-) D PCB toxicity, DHEA, antiepileptics isoflavonoids, metformin, troglitazone, trilostane HS Pregnanediol Pregnanetriol β HS HS 3 HS β β β 3 3 3 (+) Hyperadrenalism, hyperthyroidism, hyperinsulinemia, PCOS, IL-4 (+) and IL-13, IGF-1, forskolin Smoking, dioxin, isoflavones, flaxseed, procyanidins, flavonoids, EGCG, epilobium, vitamin C, anastrazole, etc; CYPc17 17,20 Lyase 17βHSD Progesterone 17-OH-Progesterone Androstenedione Testosterone chrysin, stinging nettles, (-) and other flavonoids; β 5 ketoconazole, metformin α (+) (+) 1 5 1 Sodium depletion, high prolactin More cortisol: Less cortisol: Etiocholanolone Obesity (esp. visceral), A Good insulin sensitivity, ro CAH, late-onset adrenal hyperplasia, primary adrenal CYP2 metabolic A CYP2 hyperthyroidism, ro 5 m Excess adipose, alcohol, zinc (-) insufficiency, ankylosing spondylitis, increased salt (-) syndrome, hypothyroid, m α DHT at reduced inflammation, atas a (+) deficiency, stress, cravings, resveratrol, -
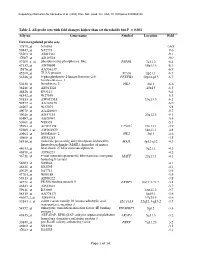
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer
ARTICLE Received 1 Nov 2013 | Accepted 17 Dec 2013 | Published 23 Jan 2014 DOI: 10.1038/ncomms4128 Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer Roland Nilsson1,2,*, Mohit Jain3,4,5,6,*,w, Nikhil Madhusudhan3,4,5, Nina Gustafsson Sheppard1,2, Laura Strittmatter3,4,5, Caroline Kampf7, Jenny Huang8, Anna Asplund7 & Vamsi K. Mootha3,4,5 Metabolic remodeling is now widely regarded as a hallmark of cancer, but it is not clear whether individual metabolic strategies are frequently exploited by many tumours. Here we compare messenger RNA profiles of 1,454 metabolic enzymes across 1,981 tumours spanning 19 cancer types to identify enzymes that are consistently differentially expressed. Our meta- analysis recovers established targets of some of the most widely used chemotherapeutics, including dihydrofolate reductase, thymidylate synthase and ribonucleotide reductase, while also spotlighting new enzymes, such as the mitochondrial proline biosynthetic enzyme PYCR1. The highest scoring pathway is mitochondrial one-carbon metabolism and is centred on MTHFD2. MTHFD2 RNA and protein are markedly elevated in many cancers and correlated with poor survival in breast cancer. MTHFD2 is expressed in the developing embryo, but is absent in most healthy adult tissues, even those that are proliferating. Our study highlights the importance of mitochondrial compartmentalization of one-carbon metabolism in cancer and raises important therapeutic hypotheses. 1 Unit of Computational Medicine, Department of Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 2 Center for Molecular Medicine, Karolinska Institutet, 17176 Stockholm, Sweden. 3 Broad Institute, Cambridge, Massachusetts 02142, USA. 4 Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, USA. -

Anabolic Steroids Detected in Bodybuilding Dietary Supplements– a Significant Risk to Public Health V
Drug Testing 1esearch article and Analysis Received; ,. June ,01$ Revised; 1 September ,01$ <ccepted: $ September ,01$ =ublished online in 7iley Online Library 3!!!.drugtestinganalysis.com4 >%I 1..1..,?dta.17,@ Anabolic steroids detected in bodybuilding dietary supplements– a significant risk to public health V. Abbate,a A. T. Kicman,b M. Evans-Brown,c J. McVeigh,d D. A. #owan,b #. Wilson,e S. J. #olese and C. J. $alkerb& Twenty-four products suspected of containing anabolic steroids and sold in fitness equipment shops in the (nited Kingdom )(K) ere analyzed for their qualitative and semi-quantitative content using full scan gas chromatography-mass spectrometry ),#-M%*, accurate mass li'uid chromatography-mass spectrometry )-#-M%*, high pressure liquid chromatography ith diode array detection )./-#-"A"*, (V-Vis, and nuclear magnetic resonance )0M1* spectroscopy. 2n addition, 3-ray crystallography enabled the identification of one of the compounds, here reference standard as not available. 4f the 56 products tested, 57 contained steroids including kno n anabolic agents8 9: of these contained steroids that ere different to those indicated on the packaging and one product contained no steroid at all. 4verall, 97 different steroids ere identified8 95 of these are controlled in the (K under the Misuse of "rugs Act 9;<9. %everal of the products contained steroids that may be considered to have considerable pharmaco- logical activity, based on their chemical structures and the amounts present. This could un ittingly e=pose users to a significant risk to their health, hich is of particular concern for na>ve users. #opyright ? 5@96 !ohn $iley A %ons, -td.