Chemistry Structure and Properties
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding
Structure and Bonding 174 Series Editor: D.M.P. Mingos Stefanie Dehnen Editor Clusters – Contemporary Insight in Structure and Bonding 174 Structure and Bonding Series Editor: D.M.P. Mingos, Oxford, United Kingdom Editorial Board: X. Duan, Beijing, China L.H. Gade, Heidelberg, Germany Y. Lu, Urbana, IL, USA F. Neese, Mulheim€ an der Ruhr, Germany J.P. Pariente, Madrid, Spain S. Schneider, Gottingen,€ Germany D. Stalke, Go¨ttingen, Germany Aims and Scope Structure and Bonding is a publication which uniquely bridges the journal and book format. Organized into topical volumes, the series publishes in depth and critical reviews on all topics concerning structure and bonding. With over 50 years of history, the series has developed from covering theoretical methods for simple molecules to more complex systems. Topics addressed in the series now include the design and engineering of molecular solids such as molecular machines, surfaces, two dimensional materials, metal clusters and supramolecular species based either on complementary hydrogen bonding networks or metal coordination centers in metal-organic framework mate- rials (MOFs). Also of interest is the study of reaction coordinates of organometallic transformations and catalytic processes, and the electronic properties of metal ions involved in important biochemical enzymatic reactions. Volumes on physical and spectroscopic techniques used to provide insights into structural and bonding problems, as well as experimental studies associated with the development of bonding models, reactivity pathways and rates of chemical processes are also relevant for the series. Structure and Bonding is able to contribute to the challenges of communicating the enormous amount of data now produced in contemporary research by producing volumes which summarize important developments in selected areas of current interest and provide the conceptual framework necessary to use and interpret mega- databases. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

Proton-Coupled Reduction of N2 Facilitated by Molecular Fe Complexes
Proton-Coupled Reduction of N2 Facilitated by Molecular Fe Complexes Thesis by Jonathan Rittle In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2016 (Defended November 9, 2015) ii 2016 Jonathan Rittle All Rights Reserved iii ACKNOWLEDGEMENTS My epochal experience in graduate school has been marked by hard work, intellectual growth, and no shortage of deadlines. These facts leave little opportunity for an acknowledgement of those whose efforts were indispensable to my progress and perseverance, and are therefore disclosed here. First and foremost, I would like to thank my advisor, Jonas Peters, for his tireless efforts in molding me into the scientist that I am today. His scientific rigor has constantly challenged me to strive for excellence and I am grateful for the knowledge and guidance he has provided. Jonas has given me the freedom to pursue scientific research of my choosing and shown a remarkable degree of patience while dealing with my belligerent tendencies. Perhaps equally important, Jonas has built a research group that is perpetually filled with the best students and postdoctoral scholars. Their contributions to my graduate experience are immeasurable and I wish them all the best of luck in their future activities. In particular, John Anderson, Dan Suess, and Ayumi Takaoka were senior graduate students who took me under their wings when I joined the lab as a naïve first-year graduate student. I am grateful for the time and effort that they collectively spent in teaching me the art of chemical synthesis and for our unforgettable experiences outside of lab. -
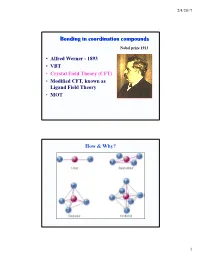
Bonding in Coordination Compounds
2/4/2017 Bonding in coordination compounds Nobel prize 1913 • Alfred Werner - 1893 • VBT • Crystal Field Theory (CFT) • Modified CFT, known as Ligand Field Theory • MOT How & Why? 1 2/4/2017 Valance Bond Theory Basic Principle A covalent bond forms when the orbtials of two atoms overlap and are occupied by a pair of electrons that have the highest probability of being located between the nuclei. Linus Carl Pauling (1901-1994) Nobel prizes: 1954, 1962 Valance Bond Model Ligand = Lewis base Metal = Lewis acid s, p and d orbitals give hybrid orbitals with specific geometries Number and type of M-L hybrid orbitals determines geometry of the complex Octahedral Complex 3+ e.g. [Cr(NH3)6] 2 2/4/2017 2- 2- Tetrahedral e.g. [Zn(OH)4] Square Planar e.g. [Ni(CN)4] Limitations of VB theory Cannot account for colour of complexes May predict magnetism wrongly Cannot account for spectrochemical series Crystal Field Theory 400 500 600 800 •The relationship between colors and complex metal ions 3 2/4/2017 Crystal Field Model A purely ionic model for transition metal complexes. Ligands are considered as point charge. Predicts the pattern of splitting of d-orbitals. Used to rationalize spectroscopic and magnetic properties. d-orbitals: look attentively along the axis Linear combination of 2 2 2 2 dz -dx and dz -dy 2 2 2 d2z -x -y 4 2/4/2017 Octahedral Field • We assume an octahedral array of negative charges placed around the metal ion (which is positive). • The ligand and orbitals lie on the same axes as negative charges. -

A TRANS INFLUENCE and Π-CONJUGATION EFFECTS on LIGAND SUBSTITUTION REACTIONS of Pt(II) COMPLEXES with TRIDENTATE PENDANT N/S-DONOR LIGANDS
Tanzania Journal of Science 44(2): 45-63, 2018 ISSN 0856-1761, e-ISSN 2507-7961 © College of Natural and Applied Sciences, University of Dar es Salaam, 2018 A TRANS INFLUENCE AND π-CONJUGATION EFFECTS ON LIGAND SUBSTITUTION REACTIONS OF Pt(II) COMPLEXES WITH TRIDENTATE PENDANT N/S-DONOR LIGANDS Grace A Kinunda Chemistry Department, University of Dar es Salaam, P. O. Box 35061, Dar es Salaam, Tanzania E-mail:[email protected]/[email protected] ABSTRACT The rate of displacement of the chloride ligands by three neutral nucleophiles (Nu) of different steric demands, namely thiourea (TU), N,N’-dimethylthiourea (DMTU) and N,N,N,’N- tetramethylthiourea (TMTU) in the complexes viz; [Pt(II)(bis(2-pyridylmethyl)amine)Cl]ClO4, (Pt1), [Pt(II){N-(2-pyridinylmethyl)-8-quinolinamine}Cl]Cl, (Pt2), [Pt(II)(bis(2- pyridylmethyl)sulfide)Cl]Cl, (Pt3) and [Pt(II){8-((2-pyridylmethyl)thiol)quinoline}Cl]Cl, (Pt4) was studied under pseudo first-order conditions as a function of concentration and temperature using a stopped-flow technique and UV-Visible spectrophotometry. The observed pseudo first- order rate constants for substitution reactions obeyed the simple rate law kobs k2Nu. The results have shown that the chloro ligand in Pt(N^S^N) complexes is more labile by two orders of magnitude than Pt(N^N^N) complexes due to the high trans labilizing effect brought by the S- donor atom. The quinoline based Pt(II) complexes (Pt2 and Pt4) have been found to be slow than their pyridine counterparts Pt1 and Pt3 due to poor π-acceptor ability of quinoline. -

1.5 Phosphines As Ligands in Transition Metal Complexes
_________________________________________________________________________Swansea University E-Theses The synthesis and characterisation of some novel compounds containing Pt-Se bonds. Webster, Christopher Alan How to cite: _________________________________________________________________________ Webster, Christopher Alan (2006) The synthesis and characterisation of some novel compounds containing Pt-Se bonds.. thesis, Swansea University. http://cronfa.swan.ac.uk/Record/cronfa43110 Use policy: _________________________________________________________________________ This item is brought to you by Swansea University. Any person downloading material is agreeing to abide by the terms of the repository licence: copies of full text items may be used or reproduced in any format or medium, without prior permission for personal research or study, educational or non-commercial purposes only. The copyright for any work remains with the original author unless otherwise specified. The full-text must not be sold in any format or medium without the formal permission of the copyright holder. Permission for multiple reproductions should be obtained from the original author. Authors are personally responsible for adhering to copyright and publisher restrictions when uploading content to the repository. Please link to the metadata record in the Swansea University repository, Cronfa (link given in the citation reference above.) http://www.swansea.ac.uk/library/researchsupport/ris-support/ The Synthesis and Characterisation of Some Novel Compounds Containing Pt-Se Bonds Christopher Alan Webster Department of Chemistry University of Wales Swansea November 2006 ProQuest Number: 10821502 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. -

Selective Additions to Unsaturated Carbon-Carbon Bonds by the Use
Florida State University Libraries Electronic Theses, Treatises and Dissertations The Graduate School 2014 Selective Additions to Unsaturated Carbon- Carbon Bonds by the Use of N-Heterocyclic Carbene-Copper(I) Catalysts Brian A. Ondrusek Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected] FLORIDA STATE UNIVERSITY COLLEGE OF ARTS AND SCIENCES SELECTIVE ADDITIONS TO UNSATURATED CARBON-CARBON BONDS BY THE USE OF N-HETEROCYCLIC CARBENE-COPPER(I) CATALYSTS By BRIAN A. ONDRUSEK A Dissertation submitted to the Department of Chemistry and Biochemistry in partial fulfillment of the requirements for the degree of Doctor of Philosophy Degree Awarded: Summer Semester, 2014 Brian Ondrusek defended this dissertation on July 1st, 2014. The members of his supervisory committee were: D. Tyler McQuade Professor Directing Dissertation Bruce Locke University Representative Michael Roper Committee Member Gregory Dudley Committee Member Igor Alabugin Committee member The Graduate School has verified and approved the above-named committee members, and certifies that the dissertation has been approved in accordance with university requirements. ii I dedicate this effort to my family, without whose support I could never have accomplished all that I have. iii ACKNOWLEDGMENTS I would like to acknowledge Dr. Tyler McQuade for taking a very green graduate student and whipping me into shape. I’m sure that at times it was no easier for him than it was for myself, but his constant support has enabled me to develop as I have into a more mature scientist. He was always willing to help me understand concepts with which I was struggling, and he was always willing to allow me to explore topics in which I was interested. -

The Synthesis and Reactivity of Heterodinuclear Iron, Ruthenium, and Cobalt Compounds Containing Bridging Dithiomethylene Ligands
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1984 The synthesis and reactivity of heterodinuclear iron, ruthenium, and cobalt compounds containing bridging dithiomethylene ligands. Attempted synthesis of the iron carbyne compound Cp(CO)Fe(triple bond)CSCH3 John R. Matachek Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Inorganic Chemistry Commons Recommended Citation Matachek, John R., "The synthesis and reactivity of heterodinuclear iron, ruthenium, and cobalt compounds containing bridging dithiomethylene ligands. Attempted synthesis of the iron carbyne compound Cp(CO)Fe(triple bond)CSCH3 " (1984). Retrospective Theses and Dissertations. 8191. https://lib.dr.iastate.edu/rtd/8191 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1.The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. -

UC Berkeley UC Berkeley Electronic Theses and Dissertations
UC Berkeley UC Berkeley Electronic Theses and Dissertations Title Design and Characterization of Catalysts for the Production of Transportation Fuels from Low Molecular Weight Alkenes Permalink https://escholarship.org/uc/item/9zp3591h Author Hanna, David Publication Date 2014 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California Design and Characterization of Catalysts for the Production of Transportation Fuels from Low Molecular Weight Alkenes by David Galil Hanna A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemical Engineering in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Alexis T. Bell, Chair Professor Roya Maboudian Professor T. Don Tilley Spring 2014 Design and Characterization of Catalysts for the Production of Transportation Fuels from Low Molecular Weight Alkenes © 2014 by David Galil Hanna Abstract Design and Characterization of Catalysts for the Production of Transportation Fuels from Low Molecular Weight Alkenes by David Galil Hanna Doctor of Philosophy in Chemical Engineering University of California, Berkeley Professor Alexis T. Bell, Chair Low molecular weight alkenes comprise a significant fraction of the effluent from fluid catalytic cracking units and of the C2-C5 hydrocarbons produced from Fischer Tropsch synthesis. Yet these products are too volatile to be blended into transportation fuel. Therefore, the aim of this research was to investigate pathways by which light alkenes could be converted to molecules with favorable gasoline or diesel characteristics. This research is relevant because such pathways would allow for the production of fuels from non-petroleum feedstocks such as biomass, coal, and natural gas. -

ABSTRACT TALIAFERRO, CHELSEA MARIE. Photophysical
ABSTRACT TALIAFERRO, CHELSEA MARIE. Photophysical Characterization and Ultrafast Dynamics of Diimine-Containing Metal Hydrides and Carbonyls (Under the direction of Prof. Felix N. Castellano). Typically the center of photochemical reactivity, the metal hydride bond is instrumental in photocatalytic reactions involving transition metal hydrides. The ability to vary between photoacidic and photohydridic character is an extraordinary trait of some transition metal hydrides, such as [Ir(Cp*)(N^N)H]+, and allows for great tuning of photocatalytic behavior. However, this complicates the mechanistic study of these reactions. Time resolved spectroscopies allow for direct observation of photoproduct generation, therefore better elucidating the process(es) by which these reactions occur in different environments. And while transient absorption spectroscopy provides invaluable information on the underlying photophysical and photochemical processes through visualization of electronic transitions of ground and excited states, specific bonds can be probed using transient infrared spectroscopy. While the Ir-H bond typically is in a clear window, far from diimine-ligand breathing modes, the low absorptivity of these modes makes probing them through time-resolved techniques difficult. To aid in the effort of observing metal hydride stretching modes via transient infrared spectroscopy, and to better understand the underlying photophysics of Ir(III) diimine-containing hydrides, a robust dihydride with a short-lived metal-to-ligand charge transfer excited was selected for photophysical characterization. The stability of this complex allowed for direct interrogation of the Ir-H vibrational stretching modes using ultrafast transient infrared spectroscopy despite the low extinction coefficients of such vibrations. The analogous deutero complex was also synthesized and photophysically characterized, producing similar conclusions. -

Open Thesis He Liu-V9.Pdf
The Pennsylvania State University The Graduate School FUNCTIONALIZATION OF TWO-DIMENSIONAL TRANSITION METAL DICHALCOGENIDES A Dissertation in Chemistry by He Liu 2021 He Liu Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2021 ii The dissertation of He Liu was reviewed and approved by the following: Mauricio Terrones Verne M. Willaman Professor of Physics Professor of Chemistry, and Materials Science & Engineering Dissertation Advisor and Chair of Committee Raymond E. Schaak DuPont Professor of Materials Chemistry and Professor of Chemical Engineering Kenneth Knappenberger Jr. Professor of Chemistry Joshua Robinson Professor of Materials Science and Engineering Philip Bevilacqua Distinguished Professor of Chemistry, Biochemistry and Molecular Biology Head of the Department of Chemistry iii ABSTRACT Chemical surface functionalization has been widely used to tune the electronic, optical, and catalytic properties of two-dimensional (2D) transition metal dichalcogenides (TMDs). This thesis focuses on applying novel chemical functionalization methods to functionalize 2D TMD materials to tune its physical and chemical properties. The controlled tuning of these properties can then be utilized to improve the performance of 2D TMD-based devices and other applications such as sensing and catalysis. In chapter one, this thesis introduces current research fronts of 2D materials including graphene, TMDs and hexagonal boron nitride (hBN). The common synthesis and functionalization methods are also categorized and discussed in this chapter. Chapters two to four focus on individual research projects where I played a leading role and are summarized below. In chapter two, we demonstrate the spontaneous chemical functionalization via coordination of Au/Ag single atoms on monolayer MoS2. -

Beyond Traditional Superatom Ligands and Cores Evan Ambrose
Beyond Traditional Superatom Ligands and Cores Evan Ambrose Doud Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2020 © 2019 Evan Ambrose Doud All Rights Reserved Abstract Beyond Traditional Superatom Ligands and Cores Evan Ambrose Doud This dissertation summarizes my research in the Roy group on the development, synthesis, and study of new N-heterocyclic carbene (NHC) based ligands and nickel phsophinidene core compositions of molecular clusters, also known as superatoms. Chapter 1 introduces superatoms as atomically precise and discreet building blocks for use in the design and synthesis of novel materials. A brief history as well as selected synthetic strategies of superatoms will be introduced. The relevant materials properties of superatoms as well as their dependence on core composition and ligand structure will be discussed. Next, the use of superatoms with specialized or functionalizable ligands to synthesize new materials will be demonstrated. This chapter details the importance of the superatom ligands and core composition is the foundation that the subsequent chapters builds upon in developing these two areas. Chapter 2 introduces a functionalized NHC as a potential superatom ligand. While not necessary for all superatom ligands, ligands that enable electronic access to the superatom core are attractive. In this chapter, the conductance of potential NHC based ligands are probed through the scanning tunneling microscope-based break-junction (STM-BJ) method. A novel method of forming single molecule junctions in situ was used and these ligands are found to display a length dependent conductance with strong coupling to the Au electrode, confirming their potential use as ligands for superatoms.